
Hematopathology — MCQs

On this page
Which one of the following lymphomas is associated with the translocation of the c-myc gene on chromosome 8?
The 'hair on end' appearance on peripheral blood smear is characteristic of which of the following conditions?
A young boy presented with dyspnea and was found to have a mediastinal mass. Which of the following conditions is known to produce mediastinal lymphadenopathy?
A 45-year-old woman has experienced worsening arthritis of her hands and feet for the past 15 years. On physical examination, there are marked deformities of the hands and feet, with ulnar deviation of the hands and swan-neck deformities of the fingers. Laboratory studies show an elevated level of rheumatoid factor. CBC shows hemoglobin, 11.6 g/dL; hematocrit, 34.8%; MCV, 87 mm3; platelet count, 268,000/ mm3; and WBC count, 6800/ mm3. There is a normal serum haptoglobin level, serum iron concentration of 20 mg/ dL, total iron-binding capacity of 195 mg/dL, percent saturation of 10.2, and serum ferritin concentration of 317 ng/mL. No fibrin split products are detected. The reticulocyte concentration is 1.1%. What is the most likely mechanism underlying this patient's hematologic abnormalities?
Spot the diagnosis?
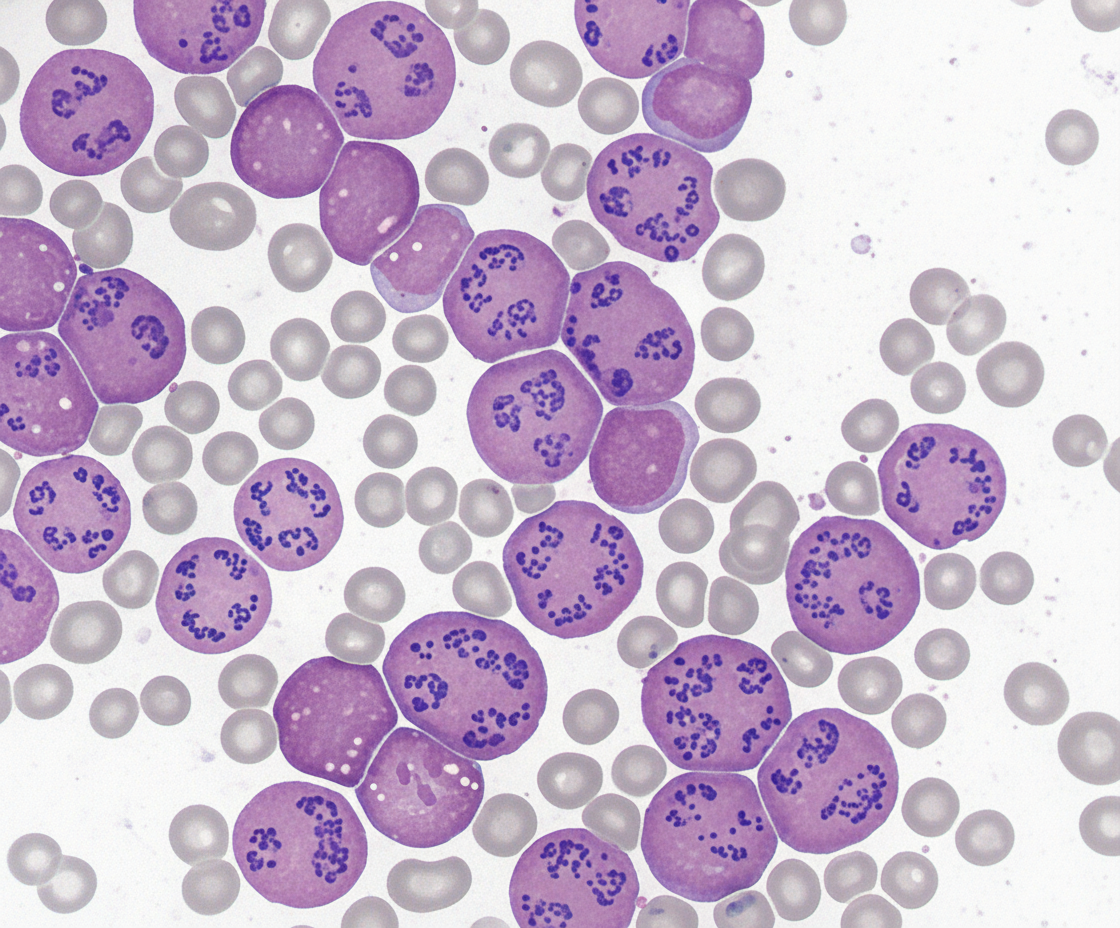
A 1-year-old girl presents with a 3-day history of fever and diarrhea. Her temperature is 38°C (101°F). A CBC reveals a normal WBC count and an increased hematocrit of 48 g/dL. What is the most likely cause of the elevated hematocrit in this patient?
Which of the following is NOT present in Sideroblastic anaemia?
Basophilic stippling is seen with which of the following conditions?
Which of the following is a consequence of extravascular hemolysis?
A 32-year-old asymptomatic female, not requiring blood transfusion, presents with Hb 13.0 gm/dl. Her HbF levels are 95% and HbA2 levels are 1.5%. Which of the following is the most likely diagnosis?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app