
Hematopathology — MCQs

On this page
What is the most malignant form of Non-Hodgkin Lymphoma (NHL)?
A leukemic patient develops disseminated intravascular coagulation. Examination of the marrow reveals hypergranular promyelocytes, some of which contain multiple Auer rods. The diagnosis of acute promyelocytic leukemia is made. Which of the following translocations is associated with the development of this disorder?
Crew haircut appearance on X-ray skull and Gandy gamma bodies are characteristic findings in which of the following conditions?
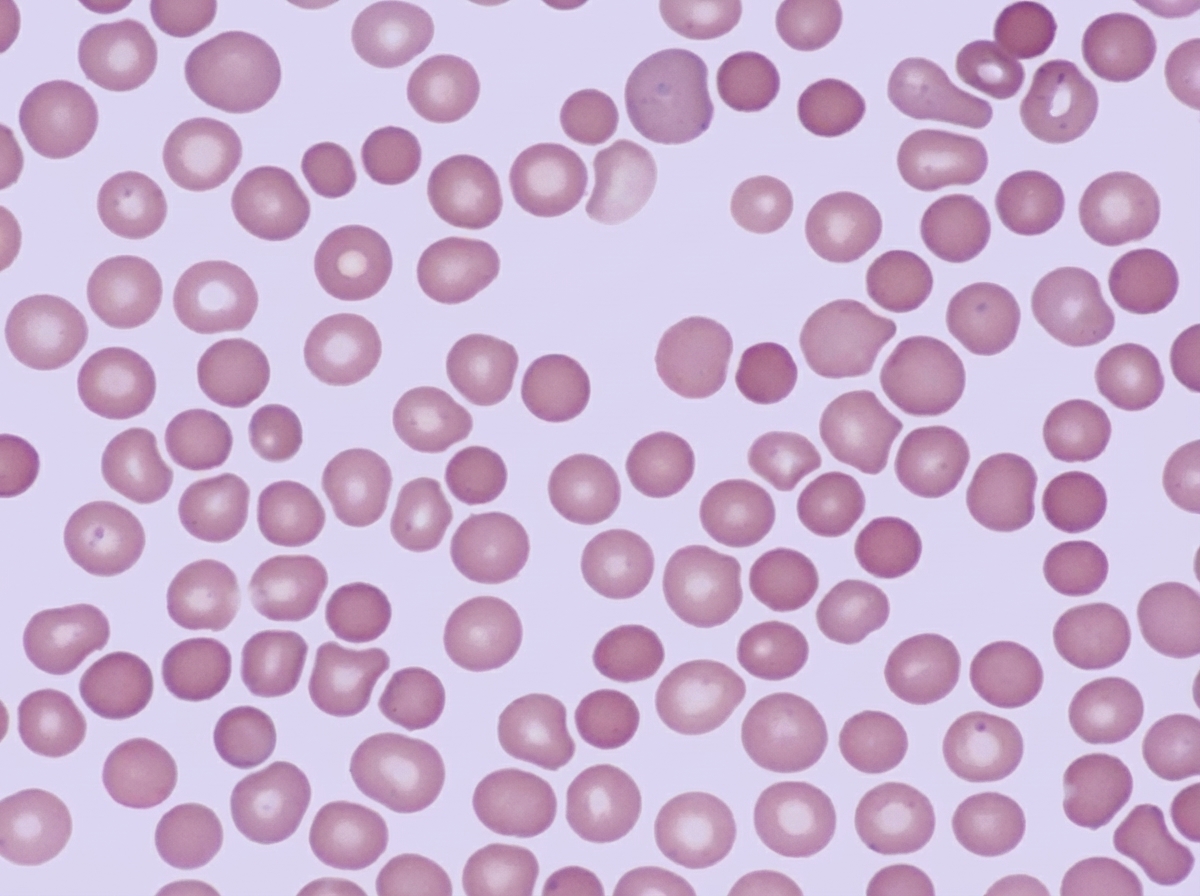
A 23-year-old man of northern European lineage presents with anemia. His father and paternal aunt had a similar illness that was treated successfully by splenectomy. His peripheral blood smear is similar to that shown in the illustration. Which of the following additional abnormalities is expected?
In sickle cell anemia, the sickle shape of red blood cells occurs due to the polymerization of which of the following?
Massive splenomegaly is not a feature of which of the following conditions?
The Ham test is primarily used to diagnose which of the following conditions?
Which of the following blood components is expected to increase in disseminated intravascular coagulation?
All of the following are true about sickle cell anemia except?
One of the following leukemias almost never develops after radiation?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app