
Hematopathology — MCQs

On this page
Which of the following is an immune response cause of anemia?
Which of the following statements is incorrect regarding Hemophilia A?
Alkaline phosphatase is specific to which of the following cells?
SAG M is an abbreviation for which of the following formulations?
A 50-year-old man presents with a pruritic rash of several years' duration, characterized by erythematous, eczematoid patches and raised plaques distributed asymmetrically over the chest and abdomen. Biopsy reveals atypical CD4+ T cells with cerebriform nuclei. Further marker studies confirm a diagnosis of mycosis fungoides. Which of the following is true of this disease?
Which of the following features may be used to differentiate hemophilia A from von Willebrand disease?
A 30-year-old male presents with cervical lymphadenopathy for 2 months. There is no past history of TB or TB contact. On examination, two lymph nodes measuring 1x1 cm are palpable, rubbery in consistency without matting. Histopathological biopsy findings were noted (image not provided, assume characteristic findings for the diagnosis). Based on the clinical presentation and likely histopathological findings, what is the most likely diagnosis among the subtypes of Hodgkin's lymphoma?
A 23-year-old male presents with fever and cervical lymphadenopathy. The peripheral smear shows a specific finding. What is the most likely diagnosis?
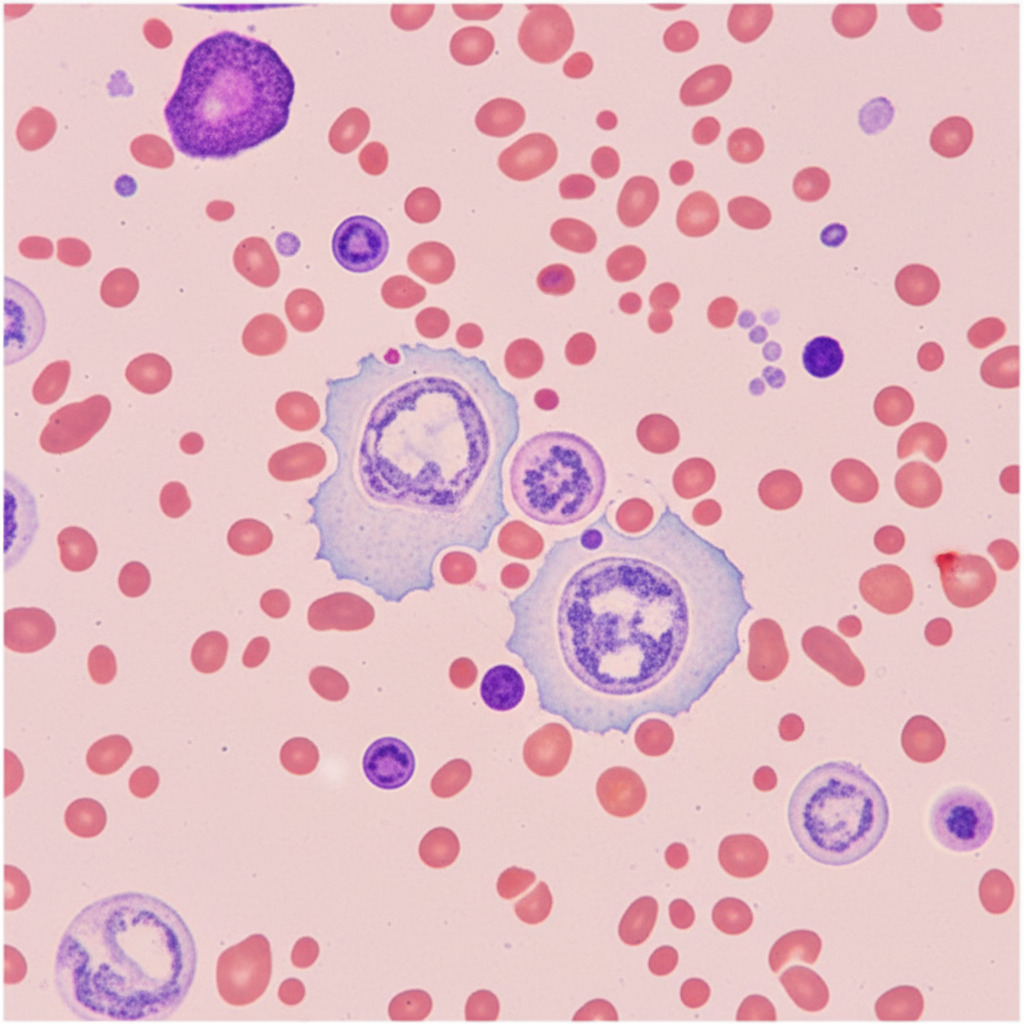
Leucopenia is not seen in which of the following conditions?
All the following conditions cause thrombocytopenia except?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app