
Hematopathology — MCQs

On this page
The genetic inheritance of Haemophilia is
Which of the following blood components has the shortest shelf life?
Consider the following statements: Haemophilia A (haemophilia) and Haemophilia B (christmas disease) 1. are variants of the same disease process 2. are due to congenital deficiency of factor VIII and factor IX respectively 3. both are sex linked characteristics and transmitted by asymptomatic females 4. can occur both in males and females Select the correct answer using the code given below:
The ideal temperature to store the whole blood in blood-bank is
A 34-year-old female presented with low hemoglobin, platelet count of 25,000 / mm³, raised PT and APTT. The image shows a peripheral smear. Which of the following fusion genes is affected?
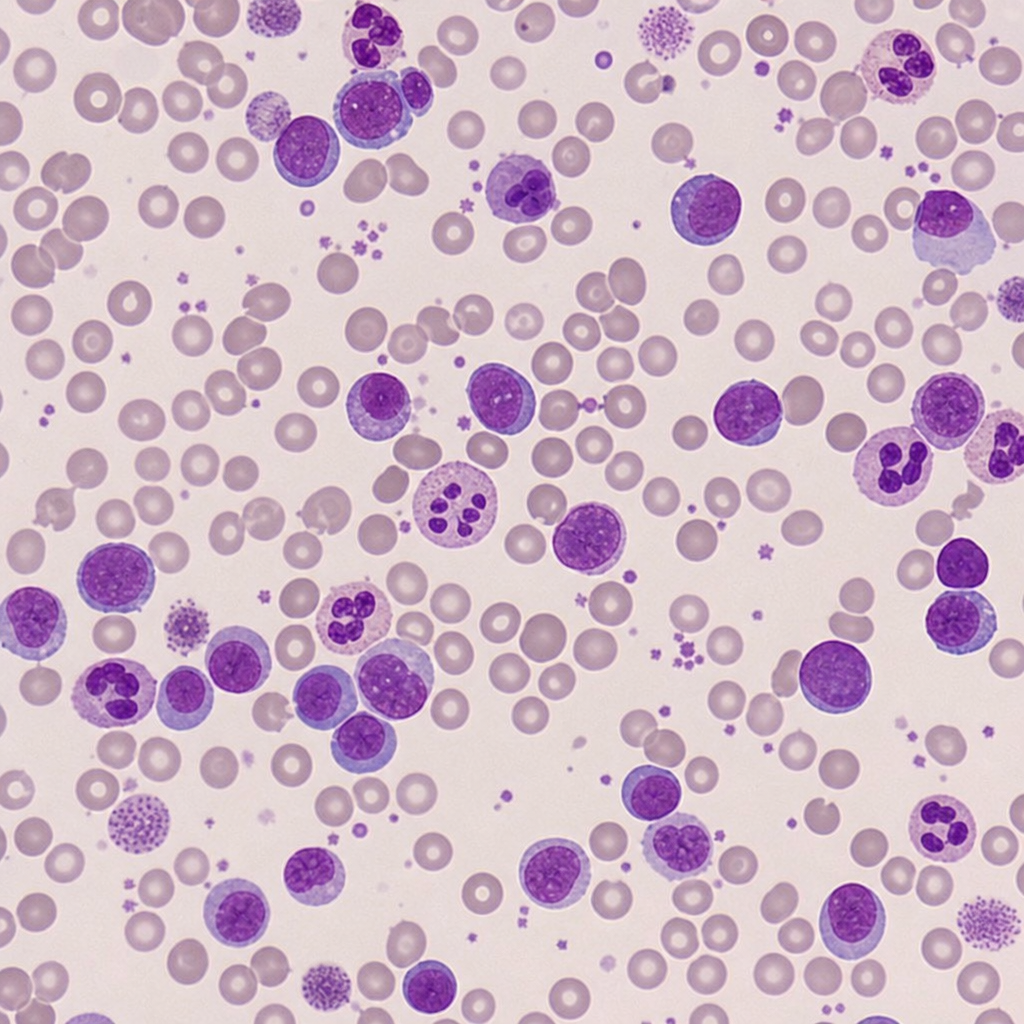
Which of the following diagnoses give the hematological picture as given below?
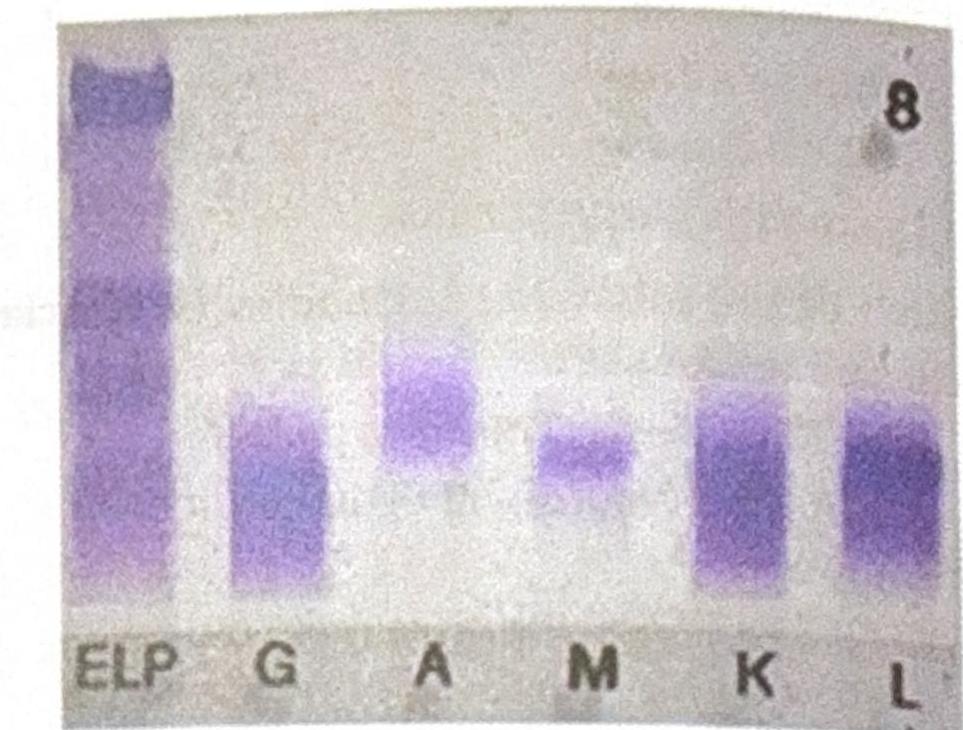
An elderly male patient presented with clinical symptoms and signs consistent with possible multiple myeloma. Electrophoresis shows an M spike, and immunofixation findings are shown below. Which of the following statements best corresponds to the findings?
The histopathology image shown is characteristic of which of the following diseases? 
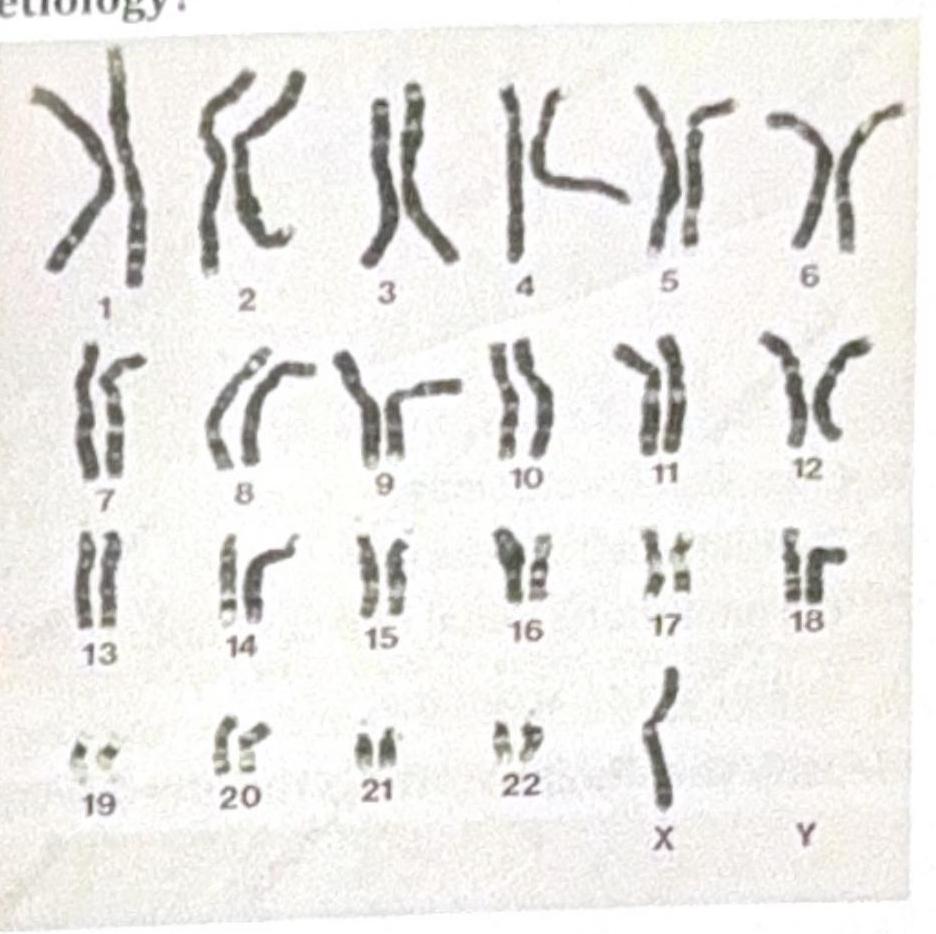
Identify the gene commonly involved in the condition shown in the image?
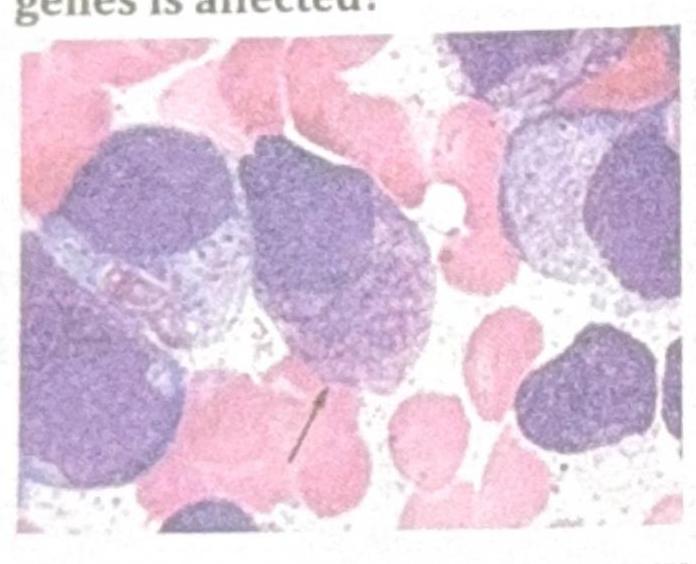
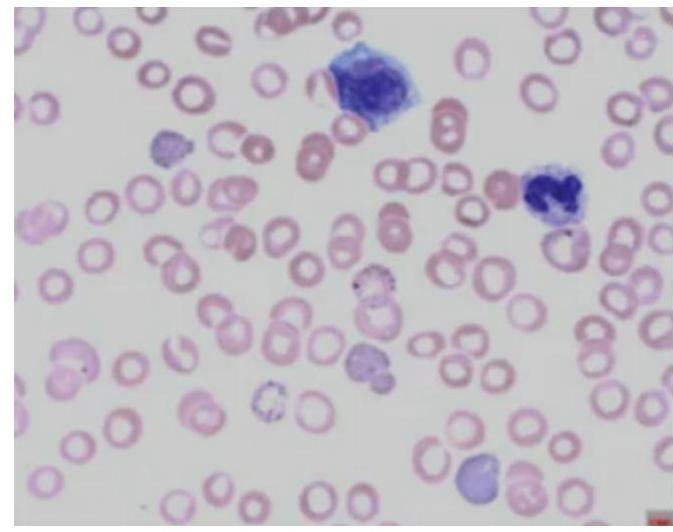
A 34-year-old female presented with low hemoglobin, platelet count of $25,000 / \mathrm{mm}^{3}$, raised PT and APTT. The image shows a peripheral smear. Which of the following fusion genes is affected?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app