
Hematopathology — MCQs

On this page
Paroxysmal nocturnal hemoglobinuria (PNH) is a disease that results from defects in which of the following?
A 10-year-old male child presents with pallor and a history of blood transfusion 2 months back. On investigation, Hb is 4.5 gms, total count is 60,000, and platelet count is 2 lakhs. Immunophenotyping shows CD10+, CD19+, CD117+, MPO+, and CD33-. What is the most probable diagnosis?
An 80-year-old asymptomatic woman on routine examination was detected to have a monoclonal spike on serum electrophoresis (IgG levels 1.5 g/dl). Bone marrow revealed 8% plasma cells. Which of the following represents the most likely diagnosis?
Which of the following conditions predisposes to leukemia?
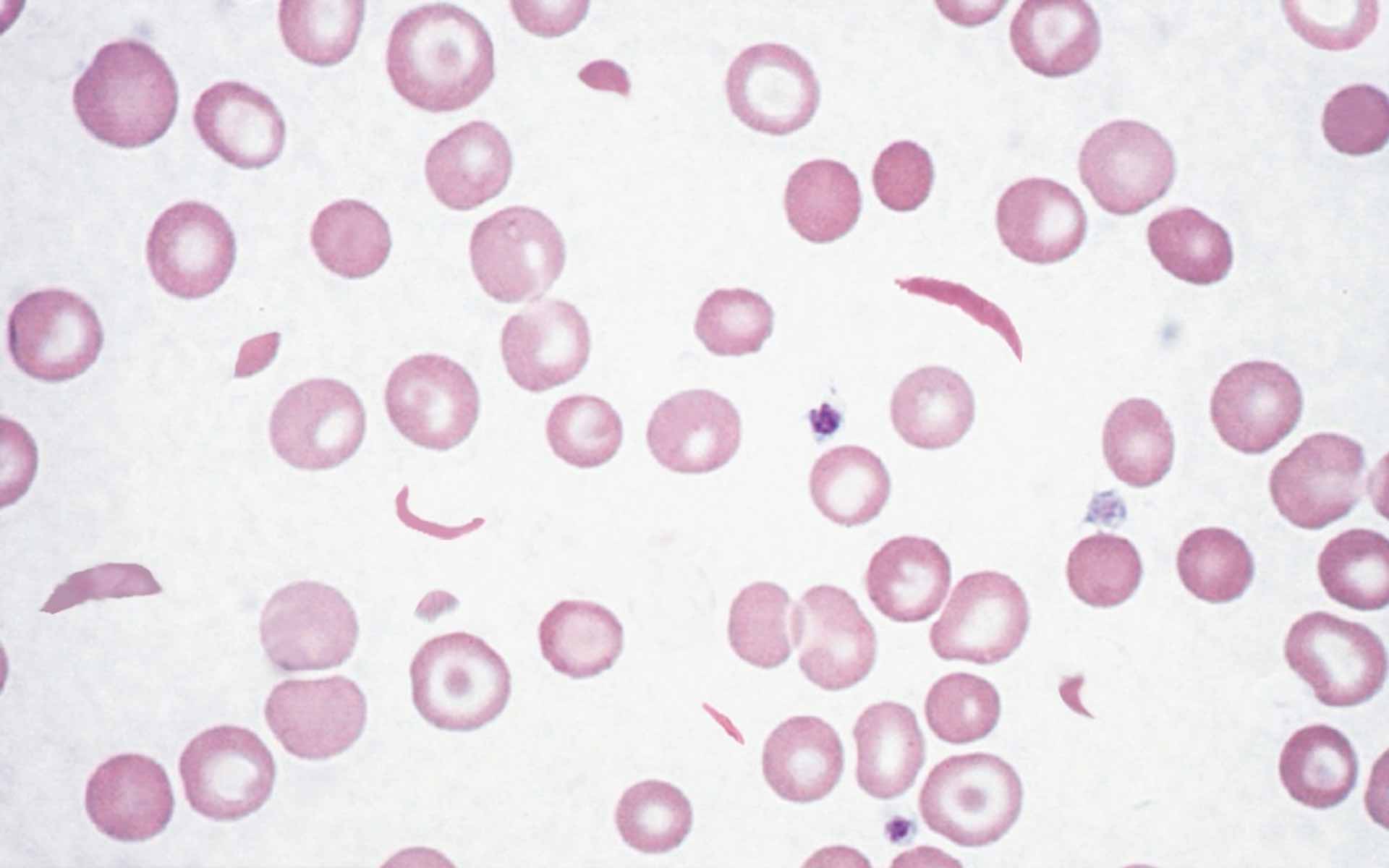
Which organ is most commonly affected in the disorder characterized by the given peripheral blood smear findings?
A 36-year-old woman presents with a 1-week history of cough and fever. On physical examination, her temperature is 38.3°C. She has diffuse crackles in all lung fields. A chest radiograph shows bilateral extensive infiltrates. Her CBC reveals a WBC count of 56,000/mm³ with 63% segmented neutrophils, 16% bands, 7% metamyelocytes, 3% myelocytes, 1% blasts, 8% lymphocytes, and 2% monocytes. A bone marrow biopsy shows normal maturation of myeloid cells. Which of the following is the most likely diagnosis?
Which cell is not seen in Hodgkin lymphoma?
Lupus anticoagulants may cause all of the following except:
Which of the viruses is not commonly implicated in the pathogenesis of Non-Hodgkin's lymphomas?
Increase in MCHC is associated with which of the following?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app