
Hematopathology — MCQs

On this page
Which of the following is NOT an autosomal recessive coagulopathy?
A 59-year-old male presents with multiple neck swellings, fever, and weight loss for 6 months. He has a history of hypertension managed with medication. General examination reveals painless lymphadenopathy in the neck, and blood investigations show anemia. Lymph node biopsy reveals cells with a delicate multilobed, 'popped corn' nucleus. Which variant of lymphoma is this?
What is the most severe form of hereditary spherocytosis caused by the mutation of?
An infant presents with mild anemia, jaundice, and splenomegaly. A complete blood count with differential reveals spherocytosis, and the reticulocyte count is elevated. The parents state that several relatives have also suffered from a similar illness. The infant's condition is most likely caused by a defect in which of the following?
The quantity of globin chain synthesis is reduced in which of the following conditions?
The histological feature shown is seen in which of the following conditions?
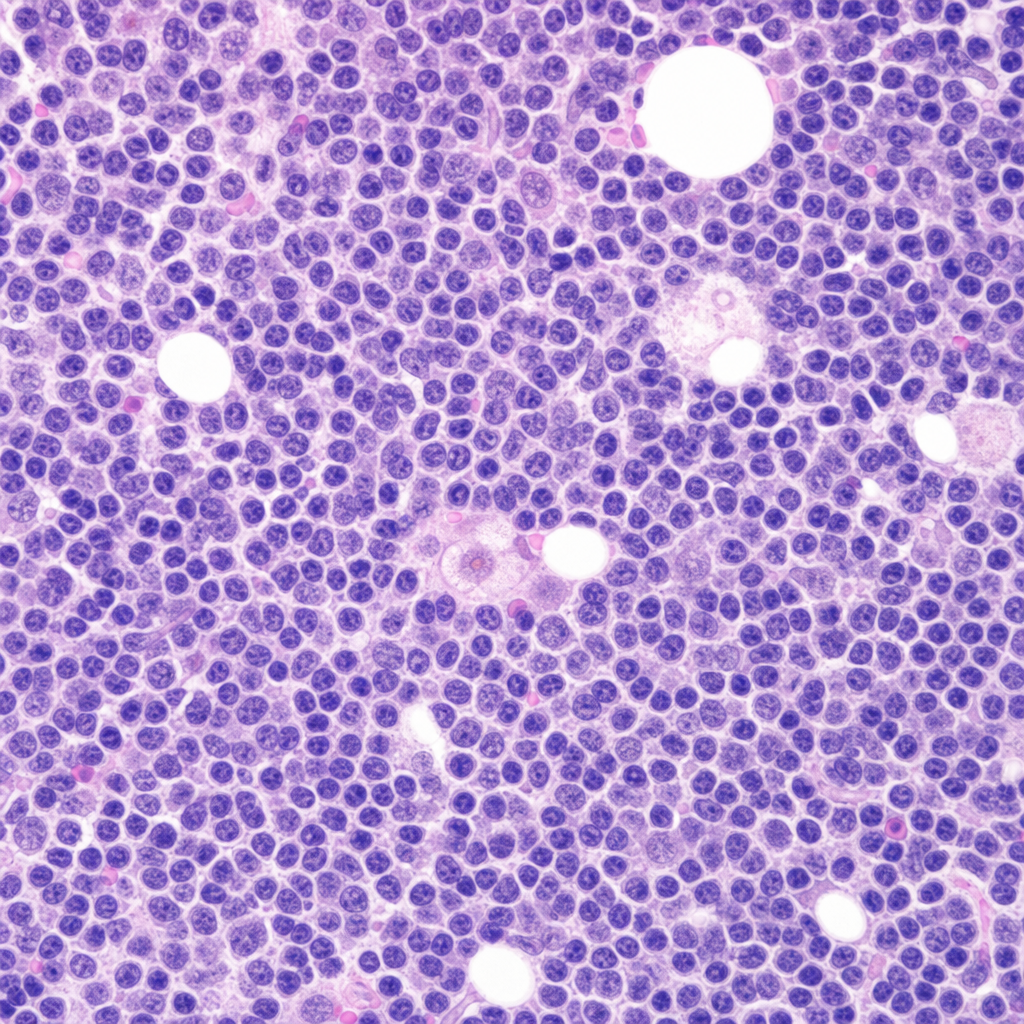
Which of the following statements regarding leukemias is true?
All are true about hemolytic disease of the newborn except?
Low LAP score may be seen in the following EXCEPT:
Which of the following toxic states is not associated with anemia?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app