
Hematopathology — MCQs

On this page
Which virus is most commonly associated with Burkitt's lymphoma?
Which of the following can cause eosinophilia?
What is the most common mutation in hemophilia?
A blood smear is best visualized at which pH?
Which of the following is not associated with the disorder characterized by findings on a peripheral blood smear?
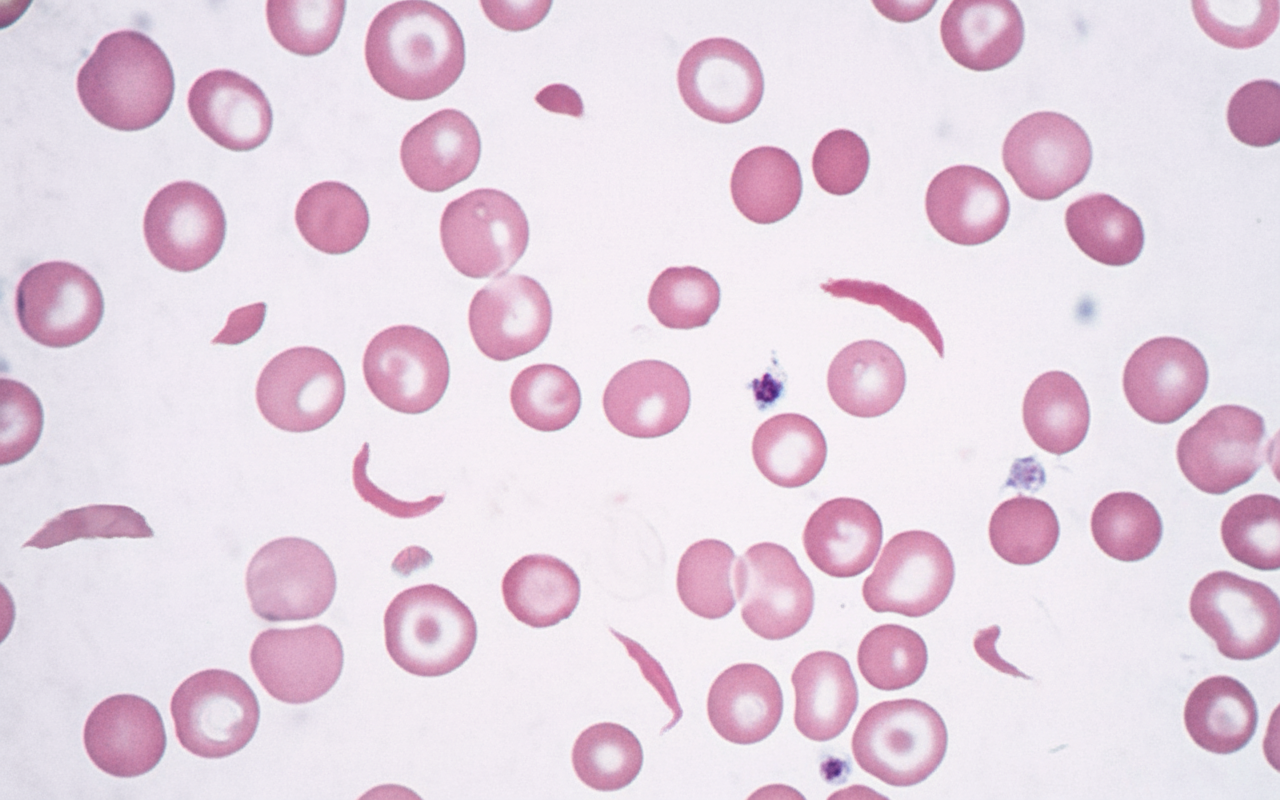
Estimation of the degradation products of the cross-linked fibrin fibers in DIC can best, specifically, and accurately be done by which of the following tests?
Which of the following statements regarding megaloblastic anemia is true?
Stored blood, which has been preserved in a blood bank, is deficient in which of the following coagulation factors?
Macrocytic anemia is not seen in which of the following conditions?
Hemolytic disease of the newborn is commonly due to incompatibility of which antigen?
Practice by Chapter
Anemias: Classification and Approach
Practice Questions
Hemolytic Anemias
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Myelodysplastic Syndromes
Practice Questions
Acute Leukemias
Practice Questions
Chronic Leukemias
Practice Questions
Lymphomas and Lymphoid Neoplasms
Practice Questions
Plasma Cell Disorders
Practice Questions
Bleeding Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app