
General Pathology — MCQs

On this page
The histological feature of shock includes:
Which of the following pigment is deposited in aging heart?
Which of the following is an example of apoptosis?
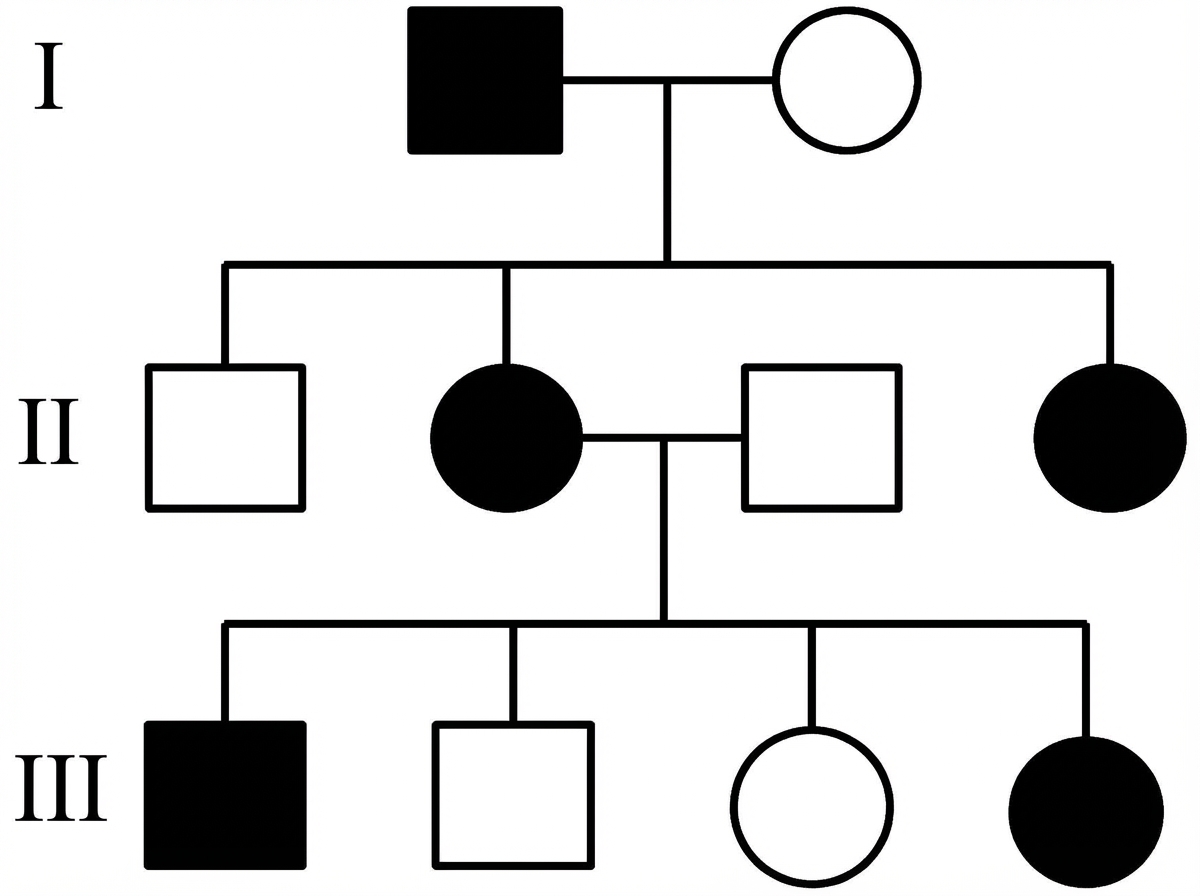
Which is the most likely inheritance pattern of the disease shown in the family pedigree? (Note: across the full extended pedigree, no male-to-male transmission has been observed.)
Telomerase activity is expressed in which of the following cell types?
A 3-year-old boy presents to the emergency room with acute distress, vague chest pain, and difficulty swallowing. He refuses to drink water. Physical examination reveals drooling and salivation. Vital signs are normal. The mother reports the child ingested a liquid drain cleaner. If this chemical was a strong acid, what histopathologic finding would be expected in the esophagus?
Decrease in cell size refers to which of the following cellular adaptations?
All of the following are TRUE regarding myoglobinuria, EXCEPT:
Which sign is most pathognomonic of irreversible cell injury?
Which of the following is the most common fixative used in electron microscopy?
Practice by Chapter
Cell Injury and Cell Death
Practice Questions
Adaptations of Cellular Growth
Practice Questions
Accumulations and Deposits
Practice Questions
Acute and Chronic Inflammation
Practice Questions
Tissue Repair and Wound Healing
Practice Questions
Hemodynamic Disorders
Practice Questions
Genetic Disorders
Practice Questions
Environmental Pathology
Practice Questions
Nutritional Diseases
Practice Questions
Molecular Basis of Disease
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app