
General Pathology — MCQs

On this page
A patient presents with hepatomegaly and joint pain. Investigations suggest abnormal intracellular accumulation of macromolecules such as lipids or glycoconjugates. Which cellular organelle is primarily responsible for this accumulation?
A child presents with hepatosplenomegaly, anemia, and bone pain. Histopathological examination reveals macrophages with a “crumpled tissue paper” appearance due to cerebroside accumulation. Deficiency of which enzyme is responsible for this condition?
Which statement correctly describes incomplete penetrance?
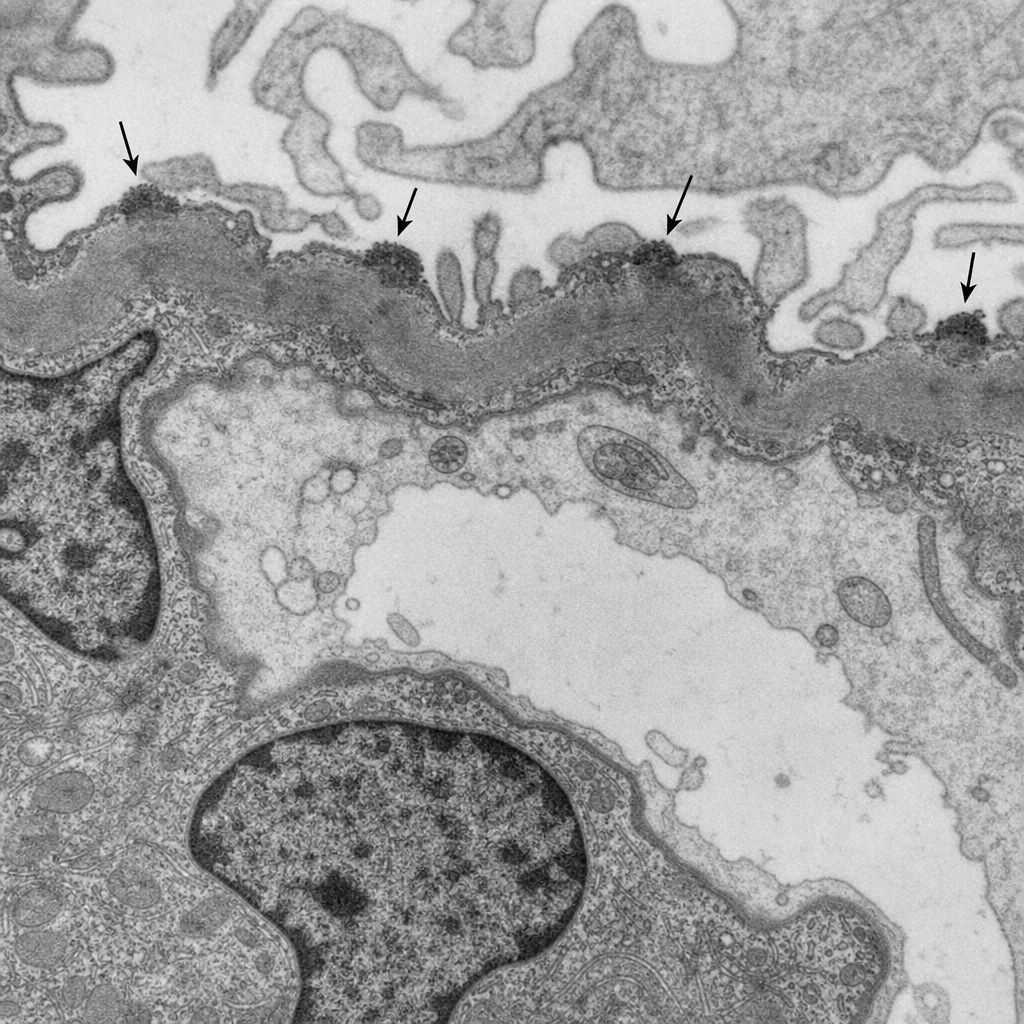
A 45-year-old male presents with nephrotic syndrome, frothy urine, and bilateral pedal edema. Serum albumin is 2.1 g/dL. Urinalysis shows 4+ proteinuria. Renal biopsy is performed. The electron microscopy image is shown (Image 1), demonstrating subepithelial electron-dense deposits along the glomerular basement membrane. Which of the following best explains the pathogenesis of the glomerular injury seen in this condition?
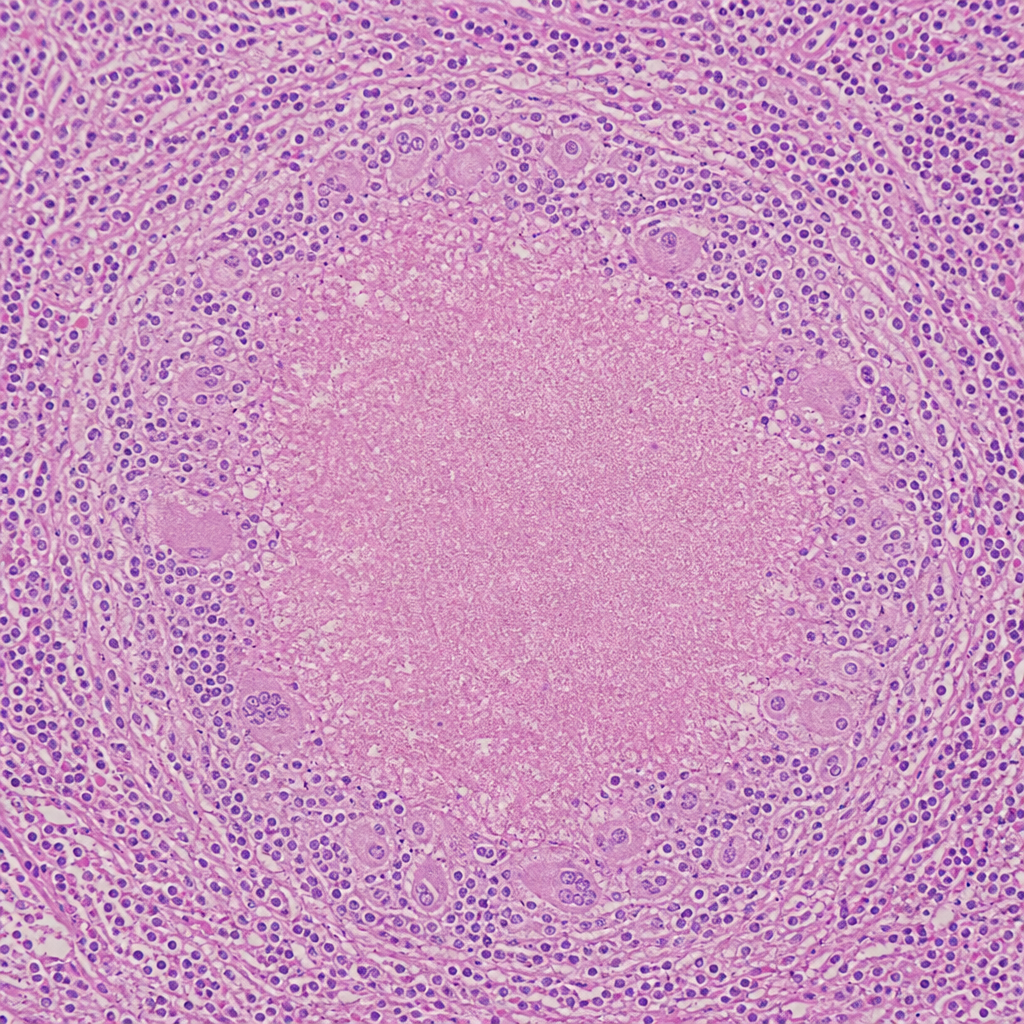
A 28-year-old male presents with low-grade fever, night sweats, weight loss, and productive cough for 8 weeks. Chest X-ray shows a right upper lobe opacity with cavitation. Sputum AFB smear is positive. The lung biopsy histology is shown in Image 3. The type of necrosis seen at the centre of the lesion in this image is also characteristically found in which of the following conditions?
A 4-year-old boy is brought to the emergency department after being found locked in a car on a summer day. His temperature is 105°F (40.6°C), he is lethargic, has decreased skin turgor, and laboratory studies show sodium 155 mEq/L, creatinine 1.8 mg/dL, and AST 250 U/L. Analyze the pathophysiological mechanism most responsible for his organ dysfunction.
Irreversible cell injury is primarily due to which of the following?
Secondary amyloidosis complicates which of the following conditions?
A 28-year-old female patient presents with patches of burns and swelling of the arm. Histopathological findings reveal swelling of the endoplasmic reticulum, blebs from the cell membrane, loss of microvilli, presence of myeloid bodies, and no changes in the nucleus. Which type of cell injury is seen in this patient?
Autopsy of the lung of an old man shows that the bronchi are lined by stratified squamous epithelium. What is this change?
Practice by Chapter
Cell Injury and Cell Death
Practice Questions
Adaptations of Cellular Growth
Practice Questions
Accumulations and Deposits
Practice Questions
Acute and Chronic Inflammation
Practice Questions
Tissue Repair and Wound Healing
Practice Questions
Hemodynamic Disorders
Practice Questions
Genetic Disorders
Practice Questions
Environmental Pathology
Practice Questions
Nutritional Diseases
Practice Questions
Molecular Basis of Disease
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app