
Endocrine Pathology — MCQs

On this page
A 45-year-old man presents with a 3 cm palpable nodule in one lobe of an otherwise normal-sized thyroid gland. Needle aspiration of the nodule demonstrates polygonal tumor cells and amyloid, but only very scanty colloid and normal follicular cells. Which of the following is the most likely diagnosis?
Ectopic ACTH syndrome is seen most commonly with which of the following conditions?
Which of the following is the most reliable feature of malignant transformation of pheochromocytoma?
A 35-year-old woman presents with a suspicious thyroid nodule on ultrasound showing microcalcifications and irregular margins. The FNAC of the lesion should reveal:
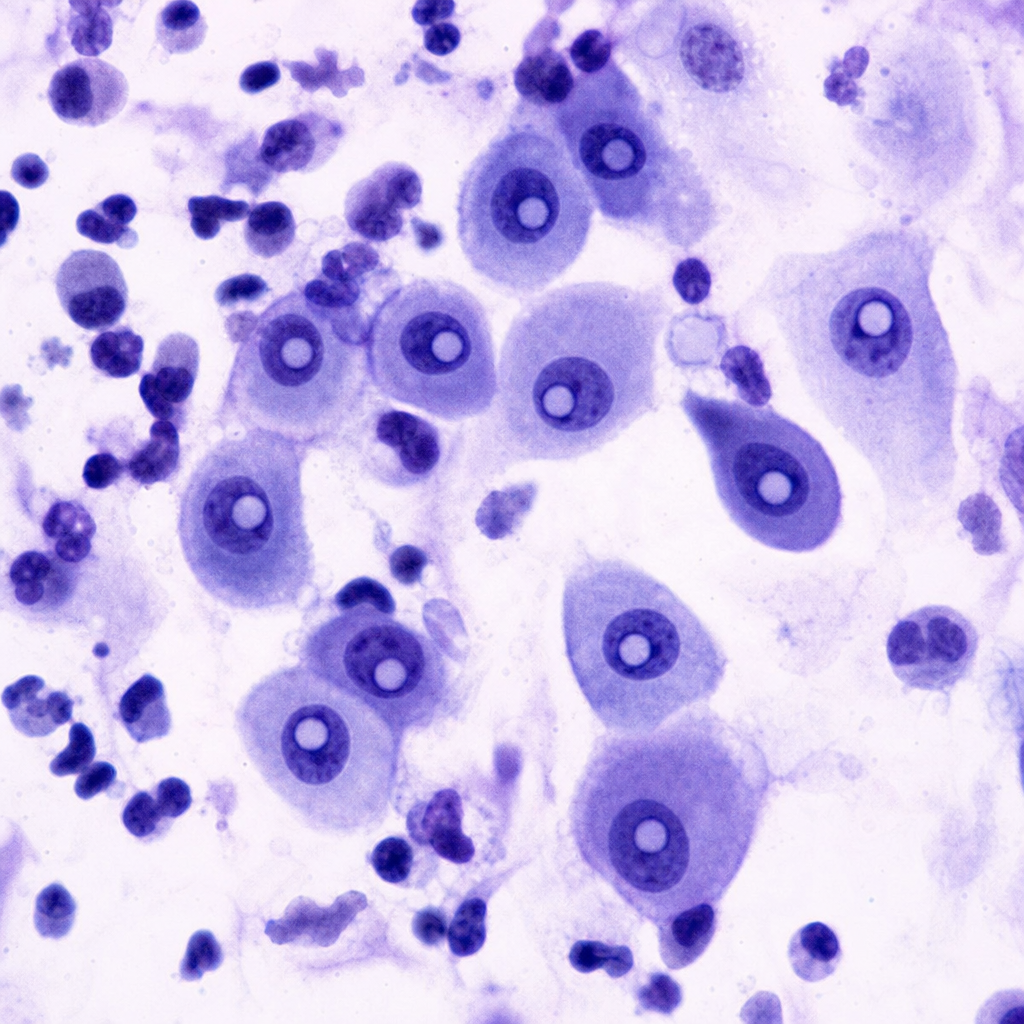
A 68-year-old man has experienced increasing malaise for 3 years. Physical examination shows no remarkable findings. Laboratory findings include a serum creatinine level of 4.9 mg/dL and a urea nitrogen level of 45 mg/dL. Abdominal CT scan shows small kidneys. Which of the following endocrine glandular lesions has developed secondary to the underlying disease in this patient?
Which of the following statements regarding Hashimoto's thyroiditis is FALSE?
Pheochromocytoma is usually associated with?
FNAC is the least diagnostic in which of the following thyroid conditions?
A 36-year-old woman presents with a nontender thyroid nodule, intermittent watery diarrhea, and a family history of thyroid cancer. A needle biopsy reveals malignant cells and homogeneous eosinophilic material. The thyroid nodule is found to be "cold" by radioiodine scintiscan. Congo red staining of the nodule reveals birefringent amyloid stroma, and genetic studies show a familial cancer syndrome. In addition to hyperparathyroidism, which of the following neoplastic diseases is this patient at risk of developing?
Microscopic examination of a specimen shows 'Psammoma bodies'. What is the most characteristic association of Psammoma bodies?
Practice by Chapter
Pituitary Gland Disorders
Practice Questions
Thyroid Gland Diseases
Practice Questions
Parathyroid Gland Pathology
Practice Questions
Adrenal Cortical Disorders
Practice Questions
Adrenal Medullary Disorders
Practice Questions
Pancreatic Endocrine Disorders
Practice Questions
Multiple Endocrine Neoplasia Syndromes
Practice Questions
Diffuse Neuroendocrine System
Practice Questions
Pineal Gland Pathology
Practice Questions
Laboratory Diagnosis of Endocrine Diseases
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app