
Endocrine Pathology — MCQs

On this page
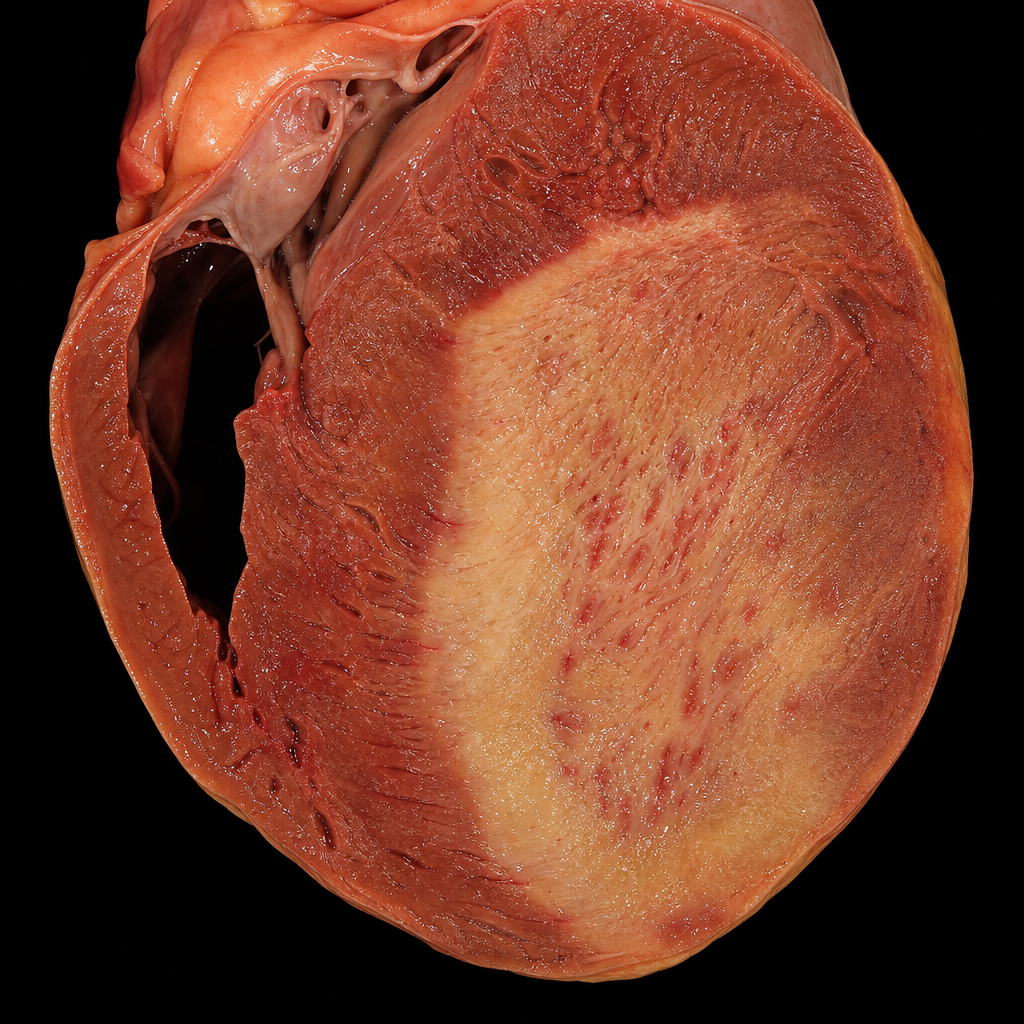
A 65-year-old male with hypertension, diabetes, and heavy smoking history is brought to the emergency department with severe crushing chest pain radiating to the left arm. He is found unresponsive 48 hours after onset of chest pain and an autopsy is performed. The gross cardiac specimen at autopsy is shown (Image 2). Which of the following cellular events is the primary pathological process responsible for the gross appearance seen in this specimen at this time point?
Which of the following mechanisms is NOT responsible for complications in Diabetes Mellitus?
Insulin non-dependent diabetes mellitus correlates with which fat reserve?
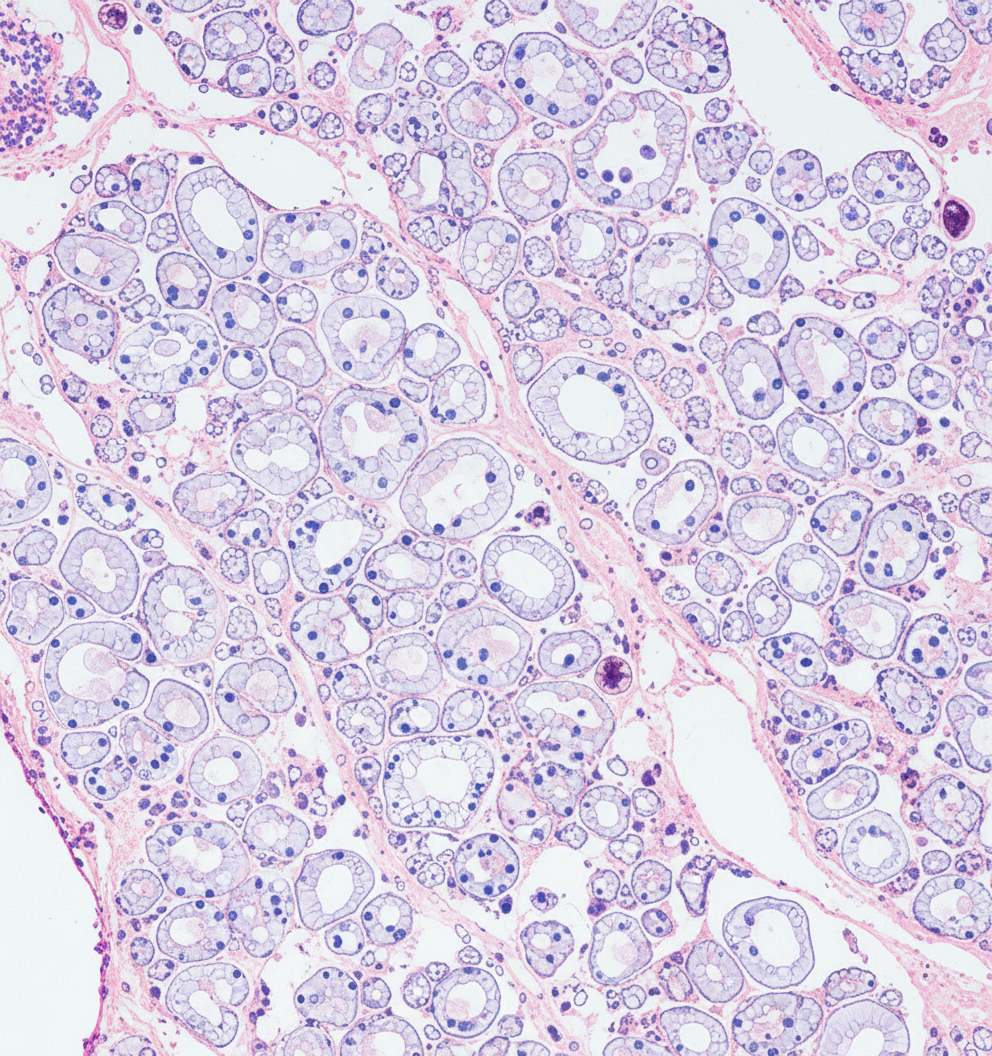
A biopsy from the parathyroid gland of a 55-year-old male, a known case of chronic kidney disease with hypertension and type II diabetes who recently developed bone pain, skin lesions, and recurrent kidney stones, is shown below. What is the most likely histopathological finding?
Osmotic damage is operative in all of the following complications of diabetes mellitus EXCEPT?
Practice by Chapter
Pituitary Gland Disorders
Practice Questions
Thyroid Gland Diseases
Practice Questions
Parathyroid Gland Pathology
Practice Questions
Adrenal Cortical Disorders
Practice Questions
Adrenal Medullary Disorders
Practice Questions
Pancreatic Endocrine Disorders
Practice Questions
Multiple Endocrine Neoplasia Syndromes
Practice Questions
Diffuse Neuroendocrine System
Practice Questions
Pineal Gland Pathology
Practice Questions
Laboratory Diagnosis of Endocrine Diseases
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app