
Bone and Soft Tissue Pathology — MCQs

On this page
Which anatomical region of the jaw bones is most likely to harbor metastatic tumors?
Punctuated lesions and floating teeth are seen in which of the following conditions?
Which of the following immunohistochemical markers would be expected to be positive in biopsies of Ewing's Sarcoma?
Which condition is characterized by the absence of dystrophin?
Mosaic pattern of lamellar bone histology is found in which of the following conditions?
Adamantinoma usually arises from?
What is the most common lesion that simulates a cementoblastoma?
Secondaries of which of the following cause osteolytic lesions except?
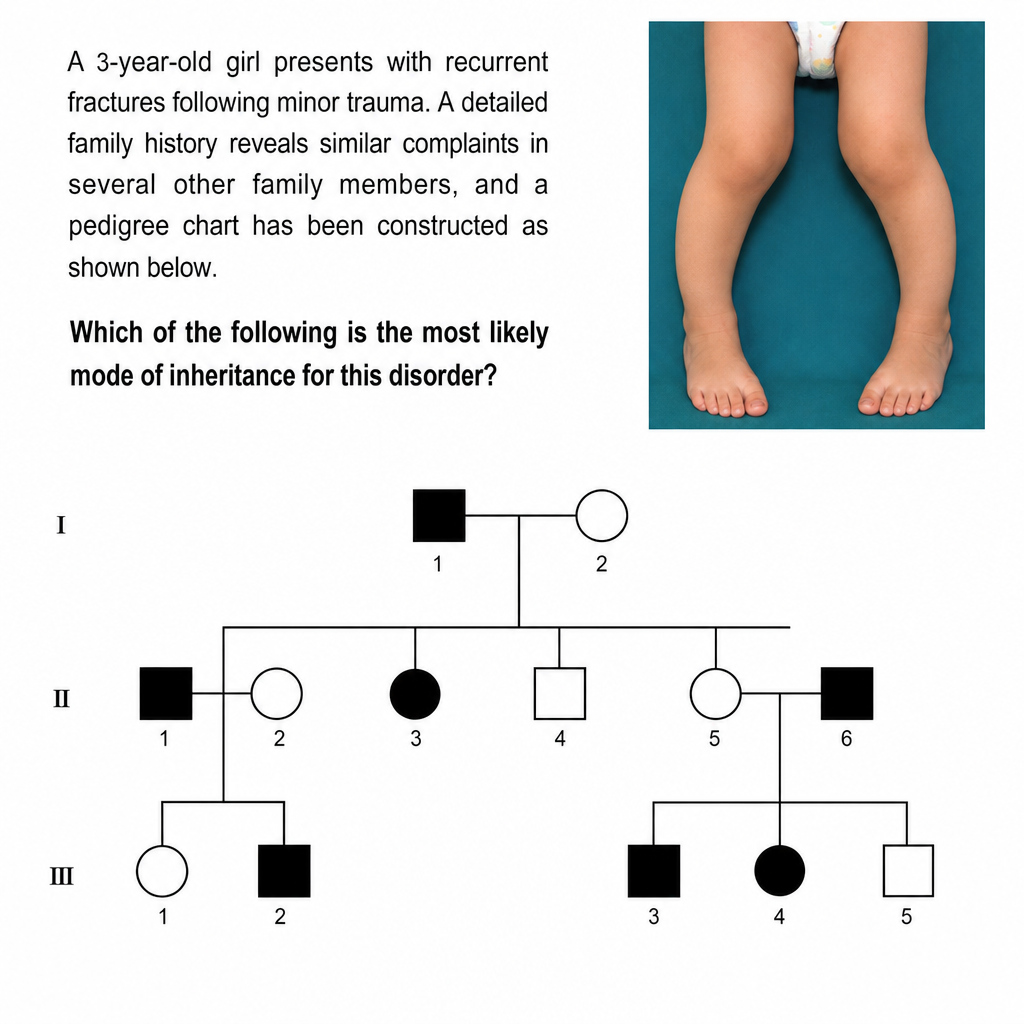
A 3-year-old girl presents with recurrent fractures following minor trauma. A detailed family history reveals similar complaints in several other family members, and a pedigree chart has been constructed (as shown below). Which of the following is the most likely mode of inheritance for this disorder?
In bone infarcts, all are true except?
Practice by Chapter
Bone Development and Growth
Practice Questions
Fracture Healing
Practice Questions
Osteomyelitis and Infectious Diseases
Practice Questions
Metabolic Bone Diseases
Practice Questions
Bone Tumors and Tumor-like Lesions
Practice Questions
Joints and Rheumatologic Diseases
Practice Questions
Soft Tissue Tumors
Practice Questions
Muscular Dystrophies and Myopathies
Practice Questions
Diseases of Tendons and Fascia
Practice Questions
Pathology of Orthopedic Implants
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app