
Lipid Metabolism — MCQs

On this page
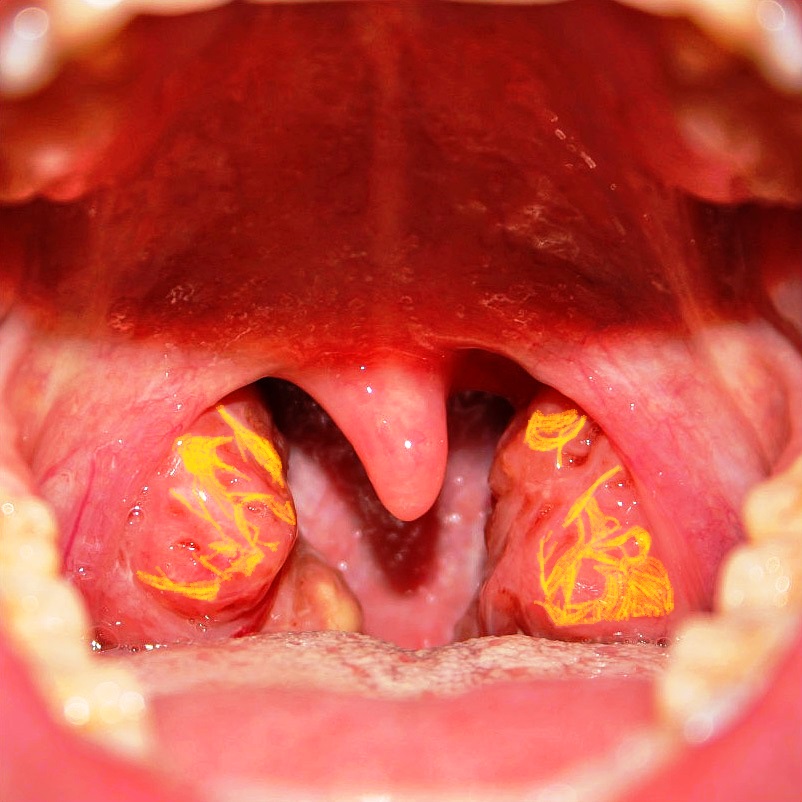
A patient presented with reduced levels of high-density lipoprotein, and ABCA1 mutation. On examination, tonsils appeared as shown in the image. What is the diagnosis?
Cyclooxygenase plays a role in which pathway?
Liver produces ketones but cannot use it due to the deficiency of which of the following enzyme?
BARTH syndrome is caused by deficiency of?
Energy reserve of the body is:
Which of the following is cardio protective?
Essential fatty acid:
Colipase is found in?
Apoprotein for chylomicron remnants:
Pregnenolone is not in the biosynthetic pathway of which substance?
Practice by Chapter
Lipid Classification and Chemistry
Practice Questions
Fatty Acid Oxidation
Practice Questions
Ketone Body Metabolism
Practice Questions
Fatty Acid Synthesis
Practice Questions
Metabolism of Triacylglycerols
Practice Questions
Phospholipid Metabolism
Practice Questions
Cholesterol Metabolism and Biosynthesis
Practice Questions
Bile Acids and Bile Salts
Practice Questions
Lipoprotein Metabolism and Transport
Practice Questions
Dyslipidemias and Atherosclerosis
Practice Questions
Prostaglandins and Eicosanoids
Practice Questions
Fatty Liver and Lipotropic Factors
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app