
Biochemistry of Hemostasis — MCQs

Activated protein C inhibits the clotting mechanism by inactivating which of the following clotting factors?
Von Willebrand disease involves a deficiency of which factor?
Which of the following statements is true regarding von Willebrand disease?
A patient on low-molecular-weight heparin suddenly develops a severe hemorrhage. What test would be most useful to assess the degree of anticoagulation?
Which of the following clotting factors in a patient on Warfarin therapy would show the earliest decrease in functional activity?
Which of the following is not typically seen in Disseminated Intravascular Coagulation (DIC)?
A 34-year-old, G1P0, presents for genetic counseling at 12 weeks' gestation. The patient has two sisters and a brother; her father has hemophilia. Her siblings are not affected, but she has a nephew who is affected. What is the inheritance pattern of this disorder?
Which of the following is a sex-linked disorder?
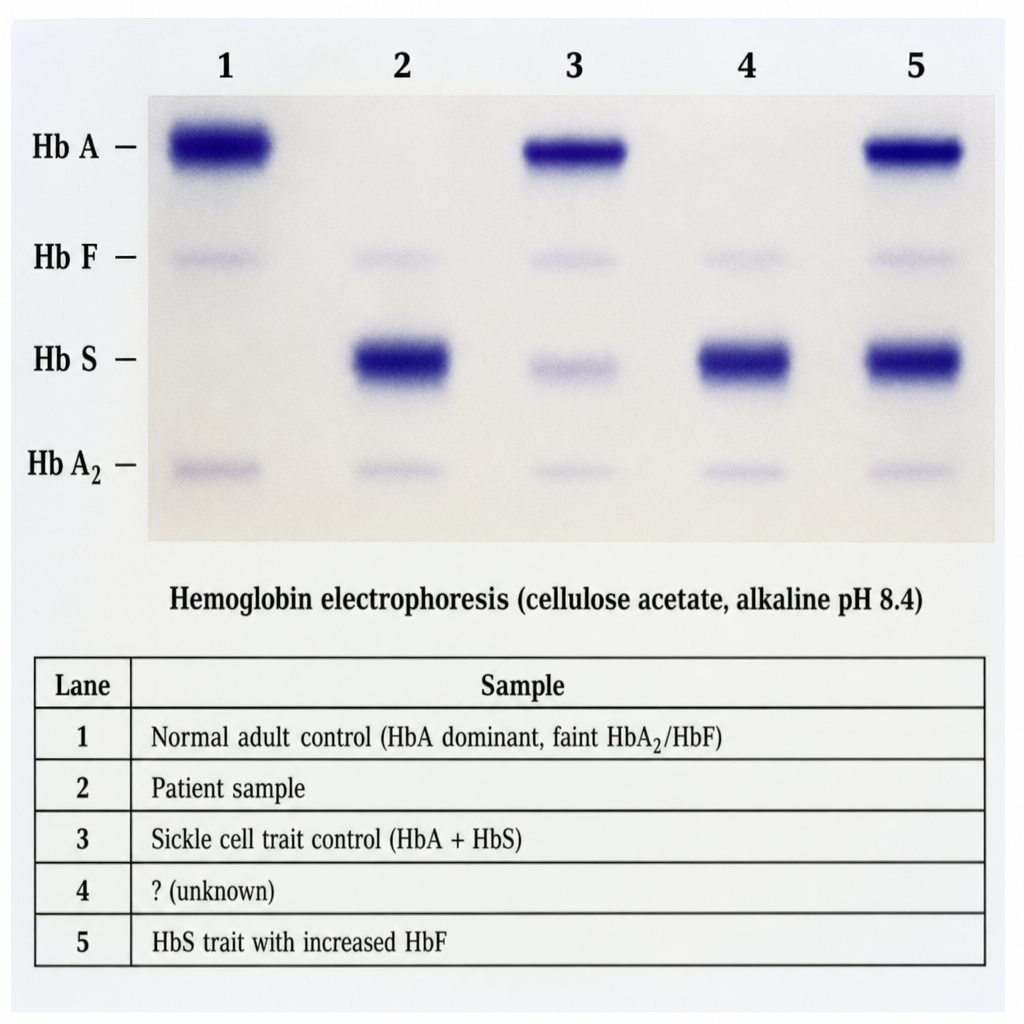
A 19-year-old male of West African descent presents with recurrent episodes of bone pain, acute chest syndrome, and a haemoglobin of 8.2 g/dL with target cells and sickle forms on peripheral smear. His parents are both clinically well. Haemoglobin electrophoresis (cellulose acetate, alkaline pH 8.4) is performed and shown in Image 1. The five lanes are: Lane 1 = Normal adult control (HbA dominant, faint HbA2/HbF); Lane 2 = Patient sample; Lane 3 = Sickle cell trait control (HbA + HbS); Lane 4 = HbC disease control (HbC only); Lane 5 = HbSC disease control (HbS + HbC, roughly equal intensity). Reference positions for HbA, HbS, HbC, and HbA2 are labeled on the gel. Lane 2 shows two bands — one co-migrating with HbS and one co-migrating with HbC — with complete absence of HbA. Based on the electrophoresis pattern in Lane 2, which combination of amino acid substitutions in the beta-globin chain is responsible for the two abnormal haemoglobin variants seen in this patient?
Ferritin biosynthesis is regulated by the serum level of which substance?
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app