
Enzyme Replacement Therapy — MCQs

Why is citrate phosphate dextrose (CPD) better than acid citrate dextrose (ACD) for storage of blood?
Which of the following is not a characteristic of Zieve syndrome?
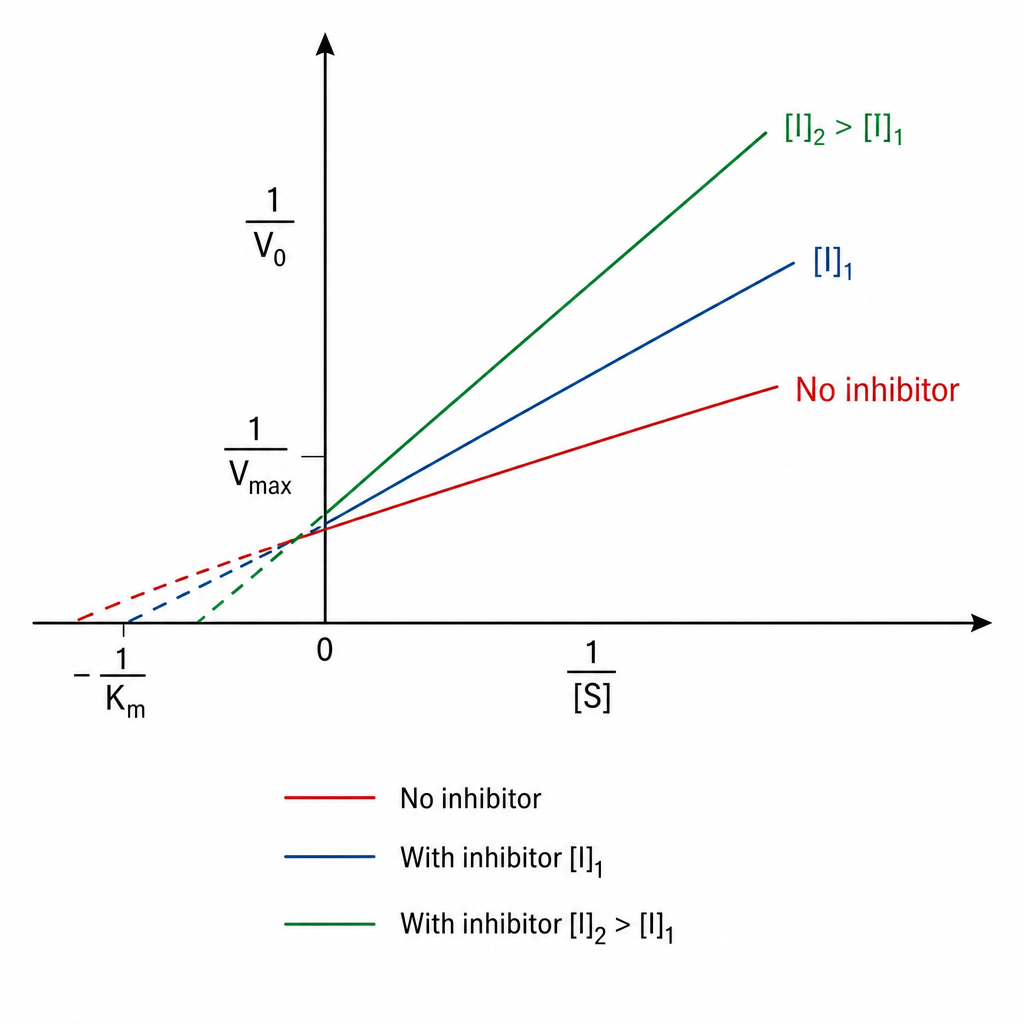
Identify the type of inhibition exhibited by A?
FALSE about Leprosy eradication programme is ?
Lysosomal transport defect is seen in which of the following conditions?
Krabbe's disease is due to deficiency of ?
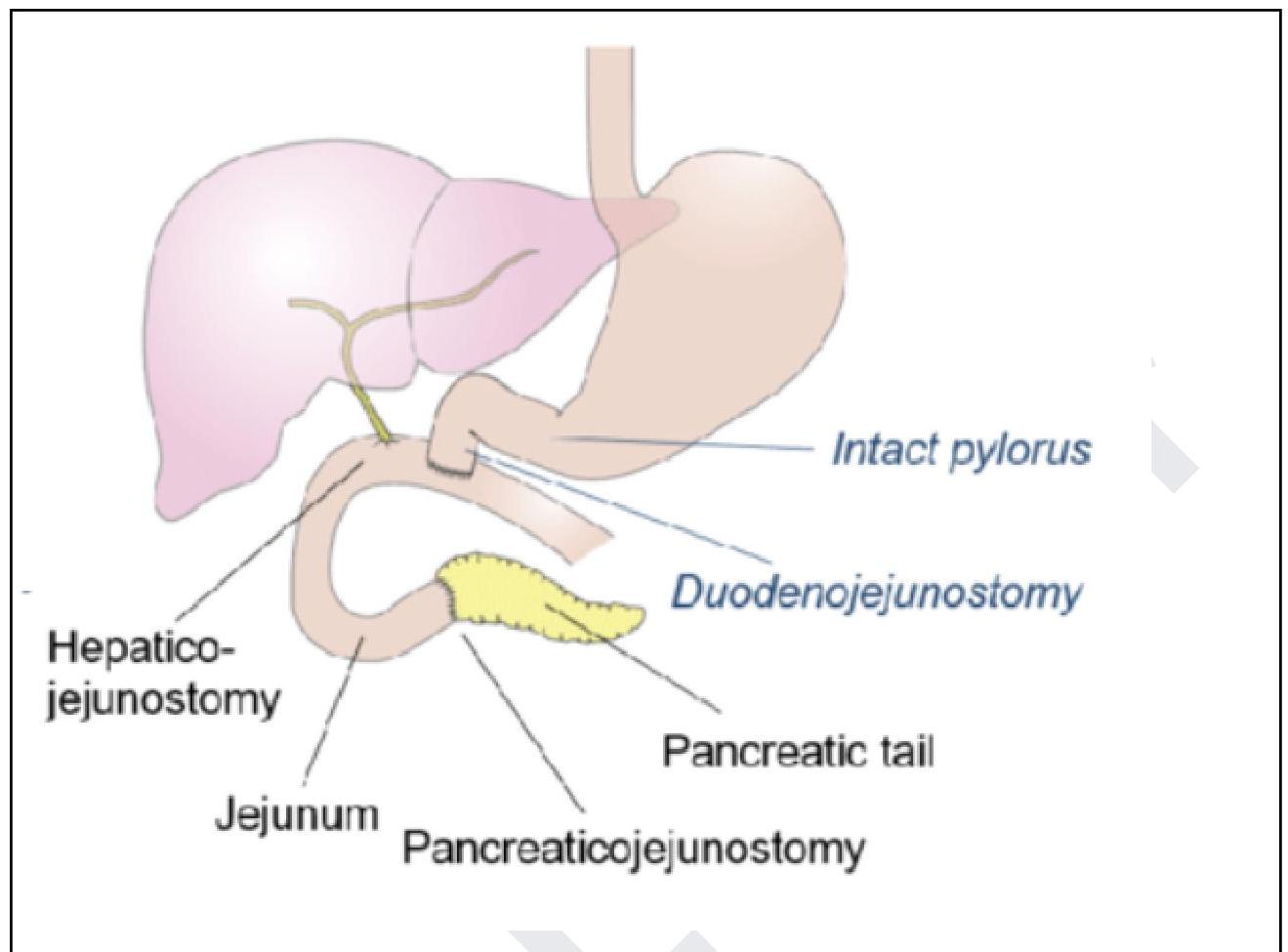
The following procedure is performed for the management of?
Splenectomy is best indicated for :
Most common deficient enzyme in Congenital adrenal hyperplasia:
An infant presented with vomiting, malnutrition, blue eyes, blonde hair & fair skin. On investigation, Guthrie test was positive. All are true regarding this disease EXCEPT:
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app