

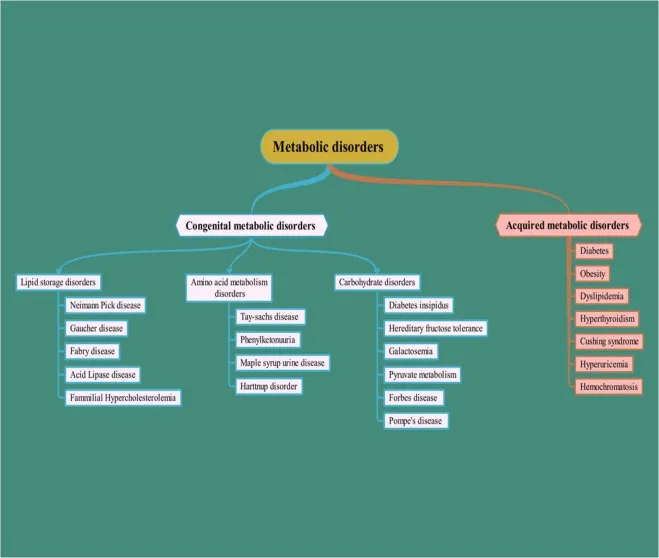
Intro & Classification - Metabolic Mayhem Map
- Inherited Metabolic Disorders (IEMs): Genetically determined biochemical pathway defects, often due to a single enzyme deficiency.
- Presentation varies: acute neonatal crisis, chronic progressive damage, or intermittent symptoms triggered by stressors (e.g., illness, fasting).
⭐ Most IEMs present in neonates after a symptom-free interval, often triggered by protein feeding (aminoacidopathies) or fasting (fatty acid oxidation defects).
Carbohydrate Disorders - Sweet Sicknesses
-
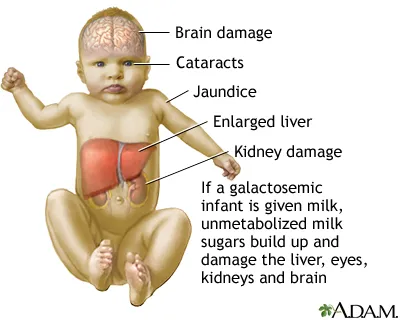
Galactosemia (Classic):
- Deficiency: Galactose-1-phosphate uridyltransferase (GALT).
- Presentation: Jaundice, hepatomegaly, infantile cataracts, intellectual disability.
- Triggers: Breast milk, standard infant formula.
- Lab: ↑ blood galactose, ↑ urine galactitol.
- 📌 Mnemonic: FAB GUT - Fructose is to Aldolase B as Galactose is to UridylTransferase.
-
Hereditary Fructose Intolerance:
- Deficiency: Aldolase B.
- Presentation: Hypoglycemia, jaundice, vomiting after consuming fructose or sucrose.
- Triggers: Fruit, honey, juice.
⭐ Neonatal sepsis with E. coli is a classic presentation of galactosemia.
- Glycogen Storage Diseases (GSDs):
- Group of disorders affecting glycogen synthesis or breakdown.
- Example: Von Gierke disease (Type I) → severe fasting hypoglycemia.
Amino Acidopathies - Protein Processing Pains
-
General: Inherited defects in amino acid metabolism leading to accumulation of toxic substrates or intermediates. Most are autosomal recessive.
-
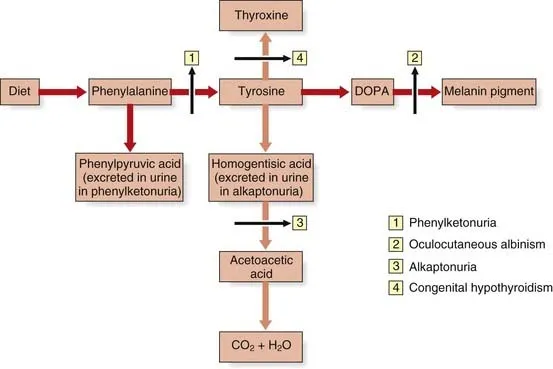
Phenylketonuria (PKU):
- Deficiency: Phenylalanine hydroxylase ($PAH$).
- Features: Musty/mousy body odor, intellectual disability, fair skin.
- Screen: Newborn screening mandatory in US.
-
Maple Syrup Urine Disease (MSUD):
- Deficiency: Branched-chain α-ketoacid dehydrogenase ($BCKDH$).
- Features: Sweet-smelling urine, neurotoxicity, poor feeding.
-
Alkaptonuria (Ochronosis):
- Deficiency: Homogentisate oxidase.
- Features: Urine turns black on standing, dark cartilage/sclerae, arthralgias.
-
Homocystinuria:
- Deficiency: Cystathionine synthase (most common).
- Features: Marfanoid habitus, ectopia lentis (downward), thrombosis.
⭐ Exam Favorite: Differentiate lens dislocation: Homocystinuria = down and inward vs. Marfan syndrome = up and outward.
Lysosomal & Lipid Storage - Cellular Clutter Crisis
- Pathophysiology: Deficient lysosomal enzymes → accumulation of undigested substrates → cellular dysfunction & organ damage.
| Disease | Deficient Enzyme | Key Features |
|---|---|---|
| Tay-Sachs | Hexosaminidase A | Cherry-red spot on macula, NO hepatosplenomegaly |
| Niemann-Pick | Sphingomyelinase | Cherry-red spot, + hepatosplenomegaly, foam cells |
| Gaucher | Glucocerebrosidase | Hepatosplenomegaly, pancytopenia, bone crises, Gaucher cells |
| Fabry | α-galactosidase A | X-linked, peripheral neuropathy, angiokeratomas |
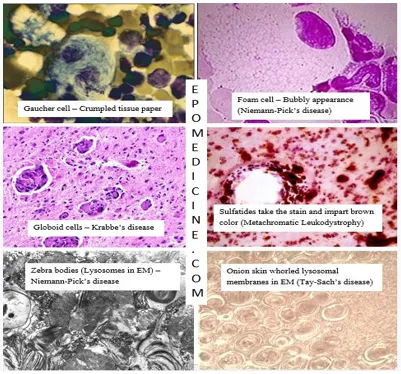
⭐ Exam Favorite: Differentiate Tay-Sachs from Niemann-Pick by organomegaly. Both present with a "cherry-red" macula, but only Niemann-Pick causes significant hepatosplenomegaly.
Mitochondrial Myopathies - Powerhouse Problems
- Genetics: Maternal inheritance only (mtDNA).
- Pathognomonic finding: "Ragged red fibers" on muscle biopsy (Gomori trichrome stain) - represent aggregates of abnormal mitochondria.
- Presentation: Variable; often myopathy, lactic acidosis, and CNS dysfunction (encephalopathy, seizures, stroke-like episodes).
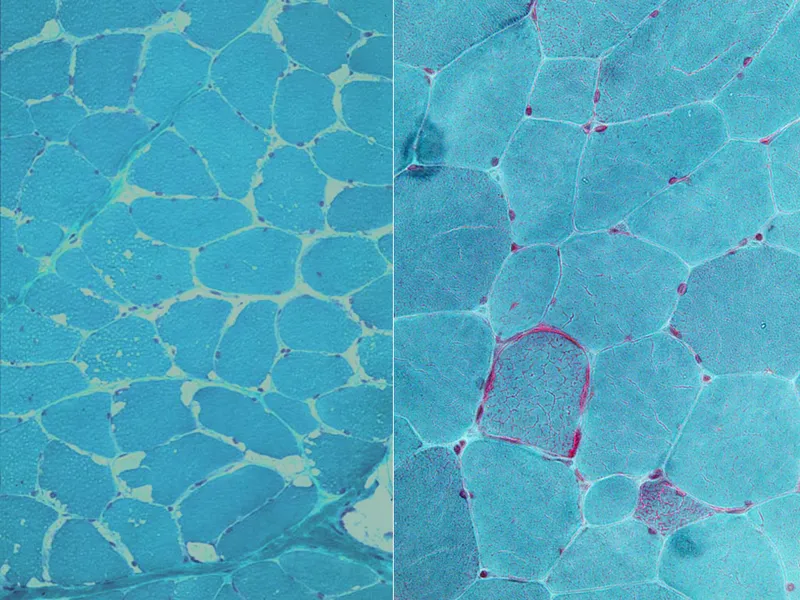
⭐ MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes) is a classic syndromic example.
High‑Yield Points - ⚡ Biggest Takeaways
- Most inherited metabolic disorders are autosomal recessive and result from enzyme deficiencies.
- Key X-linked exceptions include Fabry disease, Hunter syndrome, and G6PD deficiency.
- Presentation spans from acute neonatal crisis (vomiting, lethargy) to chronic, progressive symptoms.
- Critical lab findings often include hypoglycemia, hyperammonemia, and metabolic acidosis.
- Newborn screening is vital for early detection of conditions like PKU and galactosemia.
- Management often involves dietary restriction or enzyme replacement therapy.
Unlock the full lesson and continue reading
Signup to continue reading this lesson and unlimited access questions, flashcards, AI notes, and more