
Rheumatology and Immunology

On this page
🔬 Immunological Arsenal: Your Body's Elite Defense Network
You'll master how the immune system distinguishes self from invader, why it sometimes turns against the body's own tissues, and how to recognize and treat the resulting autoimmune and inflammatory diseases. This lesson builds your understanding from molecular pattern recognition through cellular defense mechanisms to the chemical mediators that orchestrate inflammation, then applies this foundation to diagnose rheumatologic conditions and select targeted therapies that restore immune balance. By connecting immunological principles to clinical reasoning, you'll develop the precision needed to intervene when the body's most sophisticated defense network malfunctions.
The immune system operates through two interconnected divisions: innate immunity providing immediate first-line defense within minutes, and adaptive immunity delivering specific, memory-based responses over 5-14 days. This dual-layer architecture explains why rheumatological conditions often present with both acute inflammatory features and chronic, progressive damage patterns.
📌 Remember: RAPID for immune system organization - Recognize (pattern recognition), Activate (cellular recruitment), Process (antigen presentation), Integrate (adaptive response), Destroy (target elimination). Each component operates with millisecond precision during immune surveillance.
Innate Immunity: The Immediate Response Brigade
-
Physical Barriers (First 0-30 seconds)
- Skin: 20 square feet of protective surface with pH 4.5-6.5
- Mucous membranes: 400 square meters of internal protection
- Antimicrobial peptides: >100 different defensins with broad-spectrum activity
-
Cellular Components (Minutes 1-60)
- Neutrophils: 60-70% of circulating leukocytes, 6-8 hour lifespan
- Macrophages: Tissue-resident sentinels with 48-72 hour activation
- Natural Killer cells: 5-15% of lymphocytes, direct cytotoxicity within 4 hours
- Dendritic cells: Most potent antigen presenters, 1:10,000 ratio to T cells
-
Molecular Recognition Systems
- Toll-like receptors: 10 functional TLRs recognizing pathogen-associated molecular patterns
- Complement cascade: >30 proteins with 3 activation pathways
- Cytokine networks: >50 inflammatory mediators with picomolar sensitivity
⭐ Clinical Pearl: Innate immunity dysfunction underlies 85% of autoinflammatory syndromes. Patients with recurrent fevers, sterile inflammation, and normal adaptive immune markers suggest primary innate immune defects requiring genetic testing and IL-1 blockade therapy.
Adaptive Immunity: The Precision Strike Force
| Component | Population | Function | Memory Duration | Clinical Significance |
|---|---|---|---|---|
| CD4+ T cells | 500-1500/μL | Helper coordination | 10-15 years | HIV target, autoimmune orchestration |
| CD8+ T cells | 200-900/μL | Cytotoxic killing | Lifelong | Viral clearance, tumor surveillance |
| B cells | 100-500/μL | Antibody production | Decades | Humoral immunity, autoantibody source |
| Plasma cells | <5% of B cells | Antibody secretion | Months-years | 2000 antibodies/second production |
| Memory T cells | 40-50% of total | Rapid recall | Lifelong | Vaccine efficacy, transplant rejection |
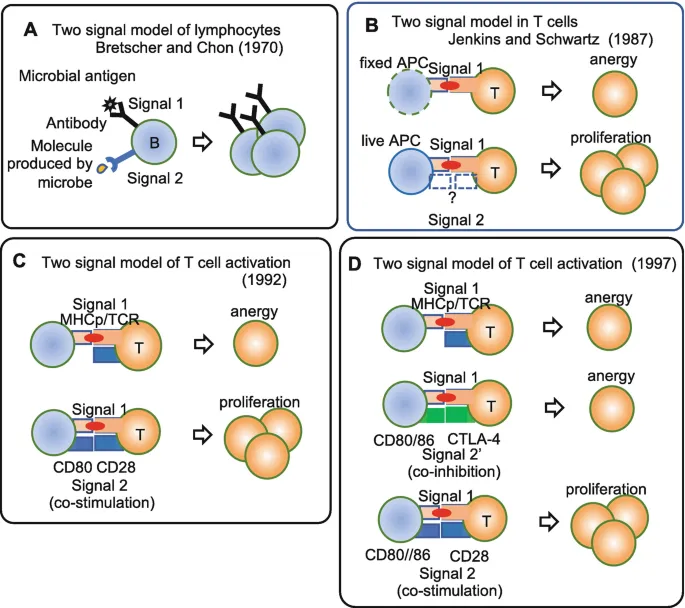
💡 Master This: T cell activation requires dual signals: Signal 1 (TCR-MHC interaction) plus Signal 2 (costimulatory molecules). Missing Signal 2 leads to anergy or deletion-the basis for checkpoint inhibitor therapy and transplant tolerance protocols.
Immunological Memory: The Learning Network
-
Primary Response Kinetics
- Lag phase: 5-10 days for initial recognition
- Peak antibody: Day 14-21 with IgM predominance
- Affinity maturation: 10-100 fold improvement over 2-3 weeks
-
Secondary Response Characteristics
- Rapid onset: 2-5 days to peak response
- Higher magnitude: 10-100 fold greater antibody levels
- Class switching: IgG, IgA, IgE predominance
- Enhanced affinity: 1000-fold improvement possible
⭐ Clinical Pearl: Immunological memory explains why live vaccines provide superior protection compared to inactivated vaccines. Memory B cells can survive >50 years, while memory T cells maintain functional responses for decades without re-exposure.
Understanding this immunological foundation reveals why rheumatological diseases follow predictable patterns: autoimmune conditions represent adaptive immunity targeting self-antigens, while autoinflammatory syndromes reflect innate immunity dysregulation. This knowledge transforms complex rheumatological presentations into logical, treatable patterns based on underlying immune mechanisms.
Connect this immunological architecture through autoimmune pathogenesis to understand how molecular mimicry, epitope spreading, and loss of tolerance create the diverse spectrum of rheumatological diseases.
🔬 Immunological Arsenal: Your Body's Elite Defense Network


⚔️ Autoimmune Warfare: When Defense Becomes Destruction
The Tolerance Breakdown: Central and Peripheral Failures
Central tolerance occurs in primary lymphoid organs where 95% of autoreactive lymphocytes undergo negative selection. Peripheral tolerance mechanisms eliminate the remaining 5% of escaped autoreactive cells through anergy, deletion, or regulatory suppression.
📌 Remember: ESCAPE mechanisms for autoimmune development - Environmental triggers (infections, drugs), Structural mimicry (molecular mimicry), Central tolerance failure (AIRE mutations), Apoptosis defects (clearance failure), Peripheral regulation loss (Treg dysfunction), Epitope spreading (tissue damage amplification).
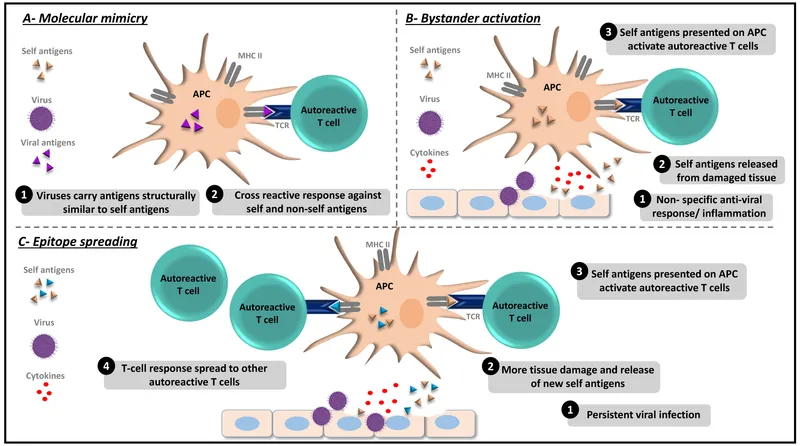
Molecular Mimicry: The Great Deception
-
Cross-Reactive Recognition Patterns
- Streptococcal M protein shares sequence homology with cardiac myosin (rheumatic fever)
- Campylobacter lipopolysaccharide mimics ganglioside GM1 (Guillain-Barré syndrome)
- Epstein-Barr virus proteins cross-react with multiple self-antigens (SLE, MS)
-
Molecular Requirements for Mimicry
- Minimum 6-8 amino acid sequence identity
- Shared conformational epitopes despite sequence differences
- HLA-restricted presentation of cross-reactive peptides
- Costimulatory signal availability during initial exposure
| Pathogen | Self-Antigen | Disease | HLA Association | Prevalence |
|---|---|---|---|---|
| Streptococcus | Cardiac myosin | Rheumatic fever | DR7, DR53 | 0.3-3% post-infection |
| Campylobacter | Ganglioside GM1 | Guillain-Barré | DQ1, DQ3 | 1:1000 infections |
| EBV | Sm antigen | SLE | DR2, DR3 | >95% SLE patients |
| Klebsiella | HLA-B27 | Ankylosing spondylitis | B27 | 90% AS patients |
| Coxsackievirus | Glutamic acid decarboxylase | Type 1 diabetes | DR3, DR4 | 5-10% genetic risk |
Epitope Spreading: The Amplification Cascade
Epitope spreading transforms localized autoimmune responses into systemic diseases through tissue damage and cryptic antigen exposure. This mechanism explains why early aggressive treatment prevents irreversible organ damage in conditions like lupus nephritis and rheumatoid arthritis.
-
Intramolecular Spreading (Weeks to months)
- Initial epitope: Single immunodominant peptide
- Tissue inflammation: Protease release and antigen processing
- Cryptic epitope exposure: Previously hidden sequences become accessible
- Response diversification: Multiple epitopes within same protein
-
Intermolecular Spreading (Months to years)
- Tissue destruction: Cell death releases intracellular antigens
- Antigen presentation: Dendritic cells process new self-proteins
- Cross-priming: T cell activation against distinct self-antigens
- Disease progression: Multi-organ involvement and systemic manifestations
💡 Master This: Epitope spreading explains why autoantibody profiles evolve over time in SLE patients. Anti-Ro/SSA may appear years before anti-dsDNA, while anti-Sm often emerges during active disease phases. Serial autoantibody monitoring predicts disease progression and organ involvement.
Regulatory T Cell Dysfunction: The Failed Peacekeepers
Regulatory T cells (Tregs) comprise 5-10% of CD4+ T cells and maintain peripheral tolerance through multiple suppressive mechanisms. Treg dysfunction underlies virtually all autoimmune diseases and represents a key therapeutic target.
-
Treg Identification Markers
- CD4+CD25+FoxP3+: Classical phenotype with >95% specificity
- CD127low: IL-7 receptor downregulation distinguishes from activated T cells
- CTLA-4high: Constitutive expression enables costimulation blockade
- Helios+: Thymic-derived versus peripheral-induced Tregs
-
Suppressive Mechanisms (Multiple pathways)
- IL-10 and TGF-β: Anti-inflammatory cytokines with local tissue effects
- CTLA-4: Competitive inhibition of CD28 costimulation
- Metabolic disruption: IL-2 consumption starves effector T cells
- Dendritic cell modulation: Reduced antigen presentation and costimulation
⭐ Clinical Pearl: Treg frequency and function predict treatment response in rheumatoid arthritis. Patients with <4% Tregs show poor response to conventional DMARDs but excellent response to IL-2 therapy which selectively expands functional Treg populations.
Understanding autoimmune pathogenesis reveals why early intervention prevents irreversible damage: epitope spreading and chronic inflammation create self-perpetuating cycles that become increasingly difficult to control. This knowledge transforms therapeutic approaches from symptom management to precision targeting of underlying immune dysfunction.
Connect these autoimmune mechanisms through inflammatory mediator networks to understand how cytokines, chemokines, and complement activation orchestrate the tissue damage patterns seen across rheumatological diseases.
⚔️ Autoimmune Warfare: When Defense Becomes Destruction


🔥 Inflammatory Mediator Networks: The Chemical Warfare System
The Cytokine Orchestra: Molecular Conductors of Inflammation
Cytokines function as molecular switches that amplify, sustain, or resolve inflammatory responses. Concentration-dependent effects mean picomolar levels trigger physiological responses, while nanomolar concentrations cause pathological inflammation.
📌 Remember: FLAMES for major inflammatory cytokines - Fever (IL-1β, TNF-α), Leukocyte recruitment (IL-8, MCP-1), Acute phase response (IL-6), Matrix destruction (MMPs), Endothelial activation (TNF-α, IL-1β), Systemic effects (cachexia, fatigue). Each operates in femtomolar to nanomolar ranges with exponential dose responses.
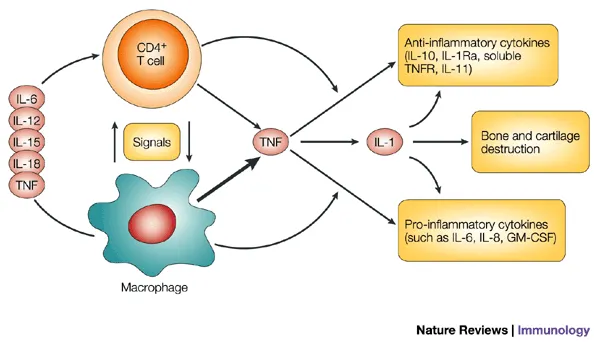
TNF-α: The Master Inflammatory Switch
Tumor Necrosis Factor-α represents the central hub of inflammatory networks, explaining why TNF inhibitors revolutionized rheumatological treatment. TNF-α triggers >200 downstream genes and coordinates virtually every aspect of acute and chronic inflammation.
-
TNF-α Biological Effects (Dose-dependent responses)
- Low doses (1-10 pg/mL): Physiological immune activation
- Moderate doses (10-100 pg/mL): Local inflammatory responses
- High doses (>100 pg/mL): Systemic inflammation and shock
- Chronic elevation: Cachexia, bone destruction, cardiovascular disease
-
Cellular Sources and Targets
- Primary producers: Macrophages (80%), T cells (15%), NK cells (5%)
- Target tissues: Endothelium, synovium, bone, liver, brain
- Receptor distribution: TNFR1 (ubiquitous), TNFR2 (immune cells)
- Half-life: 14-18 minutes in circulation, hours in tissues
| TNF Inhibitor | Mechanism | Half-life | Dosing | Efficacy (ACR20) | Infection Risk |
|---|---|---|---|---|---|
| Infliximab | Chimeric mAb | 8-10 days | IV q8 weeks | 60-70% | 2-3x baseline |
| Adalimumab | Human mAb | 14 days | SC q2 weeks | 65-75% | 2-3x baseline |
| Etanercept | Soluble receptor | 3-5 days | SC weekly | 60-65% | 1.5-2x baseline |
| Certolizumab | PEGylated Fab | 14 days | SC q2-4 weeks | 60-70% | 2-3x baseline |
| Golimumab | Human mAb | 14 days | SC monthly | 55-65% | 2-3x baseline |
Type I Interferons: The Viral Defense Gone Rogue
Type I interferons (IFN-α/β) drive lupus pathogenesis and represent emerging therapeutic targets. >50% of SLE patients show elevated interferon signatures correlating with disease activity and organ damage.
-
Interferon Signature Components
- IFI44L: Most sensitive marker with >10-fold elevation
- IFIT1: Early response gene with rapid kinetics
- ISG15: Protein modification marker of chronic activation
- MX1: Antiviral effector with sustained expression
-
Pathogenic Mechanisms in SLE
- Plasmacytoid dendritic cell activation: Immune complex uptake via TLR7/9
- Neutrophil extracellular traps: DNA-protein complexes trigger IFN production
- Autoantibody amplification: Enhanced B cell activation and class switching
- Tissue damage: Direct cytotoxic effects on kidneys, skin, CNS
💡 Master This: Interferon signature predicts lupus flares with 85% sensitivity when >2-fold elevated. Anifrolumab (anti-IFNAR1) shows greatest efficacy in high interferon signature patients with 50% reduction in flare rates and steroid-sparing effects.
The Interleukin-1 Family: Autoinflammatory Drivers
IL-1β and IL-18 drive autoinflammatory syndromes through inflammasome activation. Understanding IL-1 biology explains why anakinra and canakinumab provide dramatic responses in previously untreatable conditions.
-
Inflammasome Activation Cascade
- Signal 1: TLR activation upregulates pro-IL-1β and NLRP3
- Signal 2: Danger signals (ATP, crystals, DNA) trigger inflammasome assembly
- Caspase-1 activation: Proteolytic cleavage of pro-IL-1β to active IL-1β
- Pyroptosis: Inflammatory cell death releases additional DAMPs
-
Clinical IL-1 Targeting Strategies
- Anakinra: IL-1 receptor antagonist, daily dosing, rapid onset
- Canakinumab: Anti-IL-1β monoclonal, monthly dosing, sustained effect
- Rilonacept: Soluble IL-1 receptor, weekly dosing, broad IL-1 family inhibition
⭐ Clinical Pearl: IL-1β levels >50 pg/mL predict excellent response to IL-1 blockade in autoinflammatory syndromes. Response occurs within 24-48 hours with >90% symptom resolution in genetically confirmed cases of FMF, CAPS, and TRAPS.
Understanding inflammatory mediator networks reveals why combination therapies often succeed where monotherapy fails: redundant pathways and compensatory mechanisms require multi-target approaches. This knowledge transforms treatment from trial-and-error to precision medicine based on individual inflammatory profiles.
Connect these inflammatory networks through pattern recognition systems to understand how innate immune sensors detect tissue damage and initiate the specific inflammatory cascades that characterize different rheumatological diseases.
🔥 Inflammatory Mediator Networks: The Chemical Warfare System


🎯 Pattern Recognition Mastery: Decoding Immune Surveillance
Toll-Like Receptor Networks: The Molecular Sentinels
Toll-like receptors function as molecular switches that discriminate between pathogenic and commensal microorganisms. TLR polymorphisms explain individual susceptibility to autoimmune diseases and infection-triggered flares.
📌 Remember: NUCLEAR for endosomal TLRs - Nucleic acids (TLR3, 7, 8, 9), Unmethylated CpG (TLR9), CU-rich sequences (TLR8), Long dsRNA (TLR3), Endosomal location (acidic pH required), Autoimmune relevance (self-nucleic acid recognition), Redundant signaling (multiple TLR activation). These receptors show nanomolar sensitivity to pathogen-derived nucleic acids.
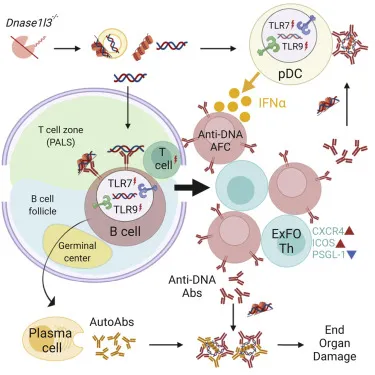
TLR7/8/9: The Nucleic Acid Detectives
Endosomal TLRs detecting nucleic acids drive lupus pathogenesis through recognition of self-DNA and self-RNA in immune complexes. This explains why antimalarial drugs (chloroquine, hydroxychloroquine) inhibit TLR signaling by raising endosomal pH.
-
TLR9 Activation in Lupus
- Self-DNA sources: Neutrophil extracellular traps, apoptotic cells, mitochondrial DNA
- Immune complex formation: Anti-dsDNA antibodies facilitate DNA uptake
- Plasmacytoid DC activation: Massive IFN-α production (>1000-fold increase)
- Positive feedback loop: IFN-α enhances TLR9 expression and sensitivity
-
TLR7/8 in RNA Recognition
- Endogenous ligands: U1-snRNP, ribosomal RNA, microRNAs
- Autoantibody complexes: Anti-Sm, anti-RNP antibodies deliver RNA to endosomes
- B cell activation: Direct TLR7 signaling promotes autoantibody production
- Therapeutic target: TLR7/8 antagonists show promise in clinical trials
| TLR | Ligand | Cell Type | Disease Association | Therapeutic Target |
|---|---|---|---|---|
| TLR3 | dsRNA | Dendritic cells | Sjögren's syndrome | Poly(I:C) inhibitors |
| TLR7 | ssRNA | B cells, pDCs | SLE (>80% patients) | IMO-8400 (antagonist) |
| TLR8 | ssRNA | Monocytes | RA synovitis | CU-CPT compounds |
| TLR9 | CpG DNA | B cells, pDCs | SLE (>90% patients) | Chloroquine, ODN inhibitors |
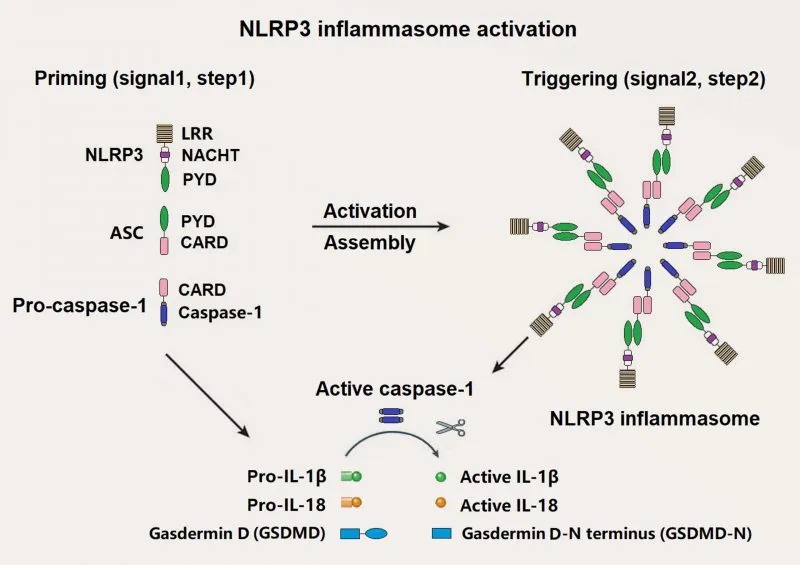
Inflammasome Complexes: The Intracellular Alarm Systems
Inflammasomes detect intracellular danger signals and activate caspase-1 to process IL-1β and IL-18. Inflammasome dysfunction underlies autoinflammatory syndromes and crystal arthropathies.
-
NLRP3 Inflammasome Activation
- Signal 1 (Priming): TLR activation upregulates NLRP3 and pro-IL-1β
- Signal 2 (Activation): K+ efflux, ROS generation, lysosomal damage
- Assembly: NLRP3-ASC-Caspase-1 complex formation
- Output: IL-1β/IL-18 release and pyroptotic cell death
-
Pathological Inflammasome Triggers
- Crystals: Uric acid (gout), calcium pyrophosphate (pseudogout), cholesterol (atherosclerosis)
- Aggregates: Amyloid-β (Alzheimer's), α-synuclein (Parkinson's)
- Metabolic stress: High glucose, fatty acids, ATP
- Genetic mutations: NLRP3, MEFV, TNFRSF1A genes
💡 Master This: Inflammasome activation requires two distinct signals separated by minutes to hours. Signal 1 (TLR priming) can occur days before Signal 2 (danger signal), explaining why infections can trigger delayed autoinflammatory flares in genetically susceptible patients.
Complement System: The Amplification Network
Complement provides rapid amplification of immune responses through >30 proteins in three convergent pathways. Complement deficiencies predispose to lupus-like syndromes, while excessive activation drives tissue damage.
-
Complement Pathway Integration
- Classical pathway: Antibody-antigen complexes (C1q, C1r, C1s)
- Lectin pathway: Mannose-binding lectin recognizes pathogen carbohydrates
- Alternative pathway: Spontaneous C3 hydrolysis with amplification loop
- Terminal pathway: C5-C9 membrane attack complex formation
-
Complement in Autoimmune Disease
- C1q deficiency: >90% develop lupus-like syndrome by age 20
- C4 deficiency: 75% lupus risk with early-onset disease
- Factor H deficiency: Atypical HUS and C3 glomerulonephritis
- C3 consumption: Active lupus nephritis marker
⭐ Clinical Pearl: Low C3/C4 levels predict lupus flares with 80% sensitivity when both decreased >25% from baseline. C3 levels <60 mg/dL and C4 <10 mg/dL indicate active nephritis requiring immediate immunosuppression and renal biopsy consideration.
Understanding pattern recognition systems reveals why certain infections trigger specific autoimmune diseases: molecular mimicry between pathogen and self-antigens combined with TLR activation breaks tolerance in genetically susceptible individuals. This knowledge enables precision prevention and targeted therapy based on individual immune profiles.
Connect these recognition systems through autoantibody development to understand how initial immune activation evolves into sustained autoimmune responses with specific antibody patterns that define distinct rheumatological diseases.
🎯 Pattern Recognition Mastery: Decoding Immune Surveillance


⚖️ Therapeutic Precision: Targeting the Immune Network
Biologic Therapy Revolution: Precision Molecular Targeting
Biologic therapies achieve target-specific inhibition with minimal off-target effects. Biomarker-guided selection improves response rates from 60-70% with empirical therapy to 85-95% with precision targeting.
📌 Remember: TARGET for biologic selection - TNF levels (TNF inhibitors), Autoantibody profile (B cell targeting), Regulatory T cell function (costimulation blockade), Genetic polymorphisms (drug metabolism), Existing infections (safety screening), Treatment history (prior failures). Each factor influences efficacy and safety with quantifiable predictive value.
TNF Inhibitor Optimization: Beyond One-Size-Fits-All
TNF inhibitors revolutionized rheumatoid arthritis treatment but show variable responses based on patient characteristics and disease phenotypes. Precision selection improves outcomes while reducing costs and adverse events.
-
Predictive Biomarkers for TNF Inhibitor Response
- Baseline TNF-α >20 pg/mL: 85% ACR20 response versus 45% with low TNF
- CRP >10 mg/L: Better response to TNF inhibitors than JAK inhibitors
- RF/ACPA positive: Superior outcomes with TNF blockade plus methotrexate
- Synovial TNF expression: High levels predict excellent response
-
TNF Inhibitor Differentiation
- Infliximab: Highest TNF binding affinity, best for severe disease
- Adalimumab: Fully human, lowest immunogenicity risk
- Etanercept: Soluble receptor, reversible binding, safest infection profile
- Certolizumab: PEGylated, no placental transfer, pregnancy option
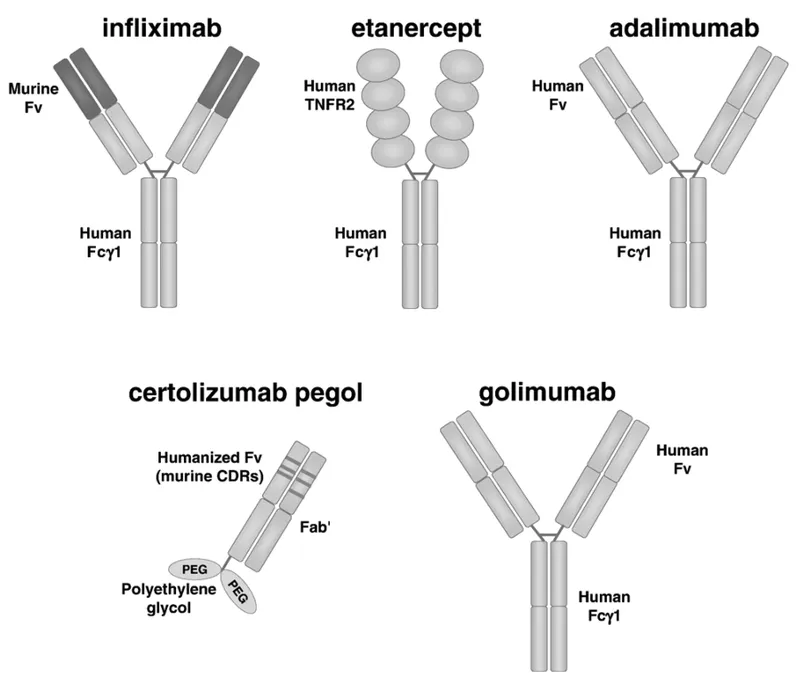
| TNF Inhibitor | Binding Affinity | Immunogenicity | Infection Risk | Pregnancy Safety | Cost (Annual) |
|---|---|---|---|---|---|
| Infliximab | Highest (Kd 0.1 nM) | 15-20% ADA | 3x baseline | Category B | $25,000 |
| Adalimumab | High (Kd 0.2 nM) | 5-10% ADA | 2.5x baseline | Category B | $60,000 |
| Etanercept | Moderate (Kd 1 nM) | <5% ADA | 1.5x baseline | Category B | $50,000 |
| Certolizumab | High (Kd 0.15 nM) | 8-12% ADA | 2x baseline | Preferred | $55,000 |
B Cell Targeting: Precision Antibody Medicine
B cell depletion and modulation provide highly effective therapy for antibody-mediated autoimmune diseases. Rituximab and newer agents offer sustained remissions with infrequent dosing.
-
B Cell Targeting Mechanisms
- Rituximab: Anti-CD20 depletes mature B cells for 6-12 months
- Belimumab: Anti-BAFF blocks B cell survival signals
- Atacicept: BAFF/APRIL inhibition targets plasma cell survival
- Obinutuzumab: Enhanced CD20 binding with improved efficacy
-
Patient Selection for B Cell Therapy
- High autoantibody titers: Anti-dsDNA >100 IU/mL, RF >100 IU/mL
- Complement consumption: Low C3/C4 indicates active immune complexes
- Organ involvement: Nephritis, CNS lupus, severe cytopenias
- Previous TNF inhibitor failure: B cell targeting often highly effective
💡 Master This: B cell depletion with rituximab provides sustained responses lasting 12-24 months after single treatment course. Peripheral B cell return occurs 6-9 months post-treatment, but clinical relapse typically lags by 6-12 additional months, allowing extended treatment-free intervals.
JAK Inhibitor Revolution: Oral Precision Medicine
Janus kinase (JAK) inhibitors provide oral alternatives to biologic therapy with rapid onset and broad anti-inflammatory effects. JAK selectivity determines efficacy and safety profiles.
-
JAK Pathway Specificity
- JAK1: IL-6, IL-10, IFN-γ signaling (inflammation, immune regulation)
- JAK2: Erythropoietin, growth hormone (hematopoiesis, metabolism)
- JAK3: IL-2, IL-4, IL-7 (T cell function, lymphocyte development)
- TYK2: IL-12, IL-23, Type I IFN (Th1/Th17 responses, antiviral immunity)
-
Clinical JAK Inhibitor Profiles
- Tofacitinib: JAK1/3 selective, excellent RA efficacy, lymphocyte effects
- Baricitinib: JAK1/2 selective, broad anti-inflammatory, anemia improvement
- Upadacitinib: JAK1 selective, highest efficacy, favorable safety
- Filgotinib: JAK1 selective, minimal JAK2 effects, preserved hematopoiesis
⭐ Clinical Pearl: JAK inhibitors show rapid onset with clinical improvement within 2-4 weeks compared to 8-12 weeks for biologics. Tofacitinib achieves ACR20 responses in 70-80% of TNF inhibitor failures, making it excellent second-line therapy for refractory RA.
Understanding therapeutic precision reveals why biomarker-guided therapy outperforms empirical treatment: individual immune profiles determine optimal targets and predict responses. This knowledge transforms rheumatology from trial-and-error to precision medicine with improved outcomes and reduced healthcare costs.
Connect these therapeutic principles through clinical integration frameworks to understand how combination strategies, treatment sequencing, and monitoring protocols optimize long-term outcomes in complex rheumatological diseases.
⚖️ Therapeutic Precision: Targeting the Immune Network


🎯 Clinical Mastery Arsenal: Rapid-Fire Diagnostic Tools
📌 Essential Arsenal: RAPID diagnostic framework - Recognize inflammatory patterns (morning stiffness >1 hour, CRP >10 mg/L), Assess autoantibody profiles (ANA patterns, specific antibodies), Pattern organ involvement (skin-joint-kidney triad), Integrate imaging findings (erosions, synovitis), Decide therapeutic targets (TNF, B cells, JAK pathways). Each component provides quantifiable diagnostic value with evidence-based thresholds.
High-Yield Diagnostic Differentiators
| Disease | Key Differentiator | Sensitivity | Specificity | Clinical Pearl |
|---|---|---|---|---|
| Rheumatoid Arthritis | ACPA + RF positive | 70% | 95% | Erosions within 2 years if untreated |
| SLE | Anti-dsDNA + low C3/C4 | 60% | 98% | Nephritis risk 80% with high titers |
| Sjögren's | Anti-Ro/SSA + Anti-La/SSB | 85% | 95% | Lymphoma risk 5% lifetime |
| Scleroderma | Anti-Scl70 or Anti-centromere | 90% | 90% | Organ involvement pattern predictor |
| Gout | Uric acid crystals + response to colchicine | 95% | 90% | Tophi develop after 10+ years |
-
TNF-α Dominant (CRP >20 mg/L, TNF >50 pg/mL)
- First-line: Adalimumab or Etanercept
- Response rate: 75-85% within 12 weeks
- Monitor: Infection screening, ADA development
-
B Cell Mediated (High autoantibodies, Low complement)
- First-line: Rituximab or Belimumab
- Response rate: 70-80% by 6 months
- Duration: 12-24 months sustained response
-
IL-1 Driven (Recurrent fevers, Sterile inflammation)
- First-line: Anakinra or Canakinumab
- Response rate: >90% within 48 hours
- Genetic testing: Confirms autoinflammatory syndrome
⭐ Master This: Early aggressive therapy within 3-6 months of symptom onset prevents irreversible joint damage in >90% of RA patients. Window of opportunity closes rapidly-erosions develop in 70% of untreated patients within 2 years, but <10% with immediate DMARD therapy.
This immunological mastery framework provides the foundation for understanding, diagnosing, and treating the full spectrum of rheumatological diseases with precision, confidence, and optimal patient outcomes.
🎯 Clinical Mastery Arsenal: Rapid-Fire Diagnostic Tools


Unlock the full lesson and continue reading
Signup to continue reading this lesson and unlimited access questions, flashcards, AI notes, and more












Have doubts about this lesson?
Ask Rezzy, your AI Study Partner, to explain anything you didn't understand
Everything you need for NEET-PG prep
Get full Oncourse access with lessons, practice questions, flashcards and AI study tools.
Scan to download app